Abstract
Moyamoya disease (MMD) is a rare, chronic, progressive intracranial arteriopathy primarily affecting children. It is characterized by stenosis or occlusion of the internal carotid and cerebral arteries, often presenting with ischemia or hemorrhagic strokes. The co-occurrence of MMD and cerebral arteriovenous malformation (AVM) is exceedingly rare. We report the case of a seven-year-old male who presented with persistent intraoral bleeding. Physical examination revealed a cavernous hemangioma in the palatal mucosa and a hyperpigmented epidermal nevus. Magnetic resonance imaging identified an intracerebral AVM, subsequently diagnosed as MMD. The patient underwent anterior interhemispheric AVM resection and received prophylactic antiplatelet therapy. He remains under clinical follow-up with no complications or neurological sequelae. Recognizing dermatological manifestations associated with MMD is essential, particularly when concurrent vascular anomalies are present. A multidisciplinary approach, incorporating both endovascular and surgical strategies, is critical in managing such complex cases.
Keywords: arteriovenous malformation, child, hemangioma, Moyamoya disease, nevus
INTRODUCTION
Moyamoya disease (MMD) is a progressive steno-occlusive disorder involving the supraclinoid segments of the internal carotid arteries and the proximal portions of the middle and anterior cerebral arteries. This condition leads to early-onset ischemic or hemorrhagic strokes. The term [ITALIC]”Moyamoya”[/ITALIC], derived from Japanese, describes the hazy, smoke-like appearance of the collateral vascular network seen on angiographic imaging. Although initially identified in East Asian populations, MMD is now recognized globally as a notable cause of pediatric stroke.1,2
MMD, a rare condition that can be idiopathic or associated with various underlying disorders, including neurofibromatosis-1, sickle cell anemia, Down’s syndrome, antiphospholipid syndrome, renal artery stenosis, congenital heart defects, and autoimmune thyroiditis. Based on the presence or absence of associated conditions, MMD is classified into two categories: idiopathic Moyamoya disease and Moyamoya syndrome (MMS). The former is diagnosed in patients without any identifiable underlying pathology, while the latter is defined by its association with systemic diseases.3-5 Although MMD primarily affects the cerebral vasculature, it may also involve other organs such as the kidneys, eyes, musculoskeletal system, and skin. Comprehensive systemic evaluation is therefore essential to identify possible syndromic associations and prevent delayed or missed diagnoses.3-5
In this report, we present the case of a seven-year-old boy who was admitted with persistent intraoral bleeding caused by a cavernous hemangioma located on the palatal mucosa. Cranial magnetic resonance imaging (MRI) revealed cerebral arteriovenous malformations (AVM) consistent with a diagnosis of Moyamoya disease.
CASE REPORT
A seven-year-old male patient was admitted with persistent oral bleeding, which originated from a hemorrhagic cavernous hemangioma located on the right side of the palatal mucosa. The bleeding was continuous and unresponsive to conservative measures.
The patient had been born via spontaneous vaginal delivery and had undergone surgery for strabismus at the age of two. There was a history of consanguinity between the parents (first-degree cousins).
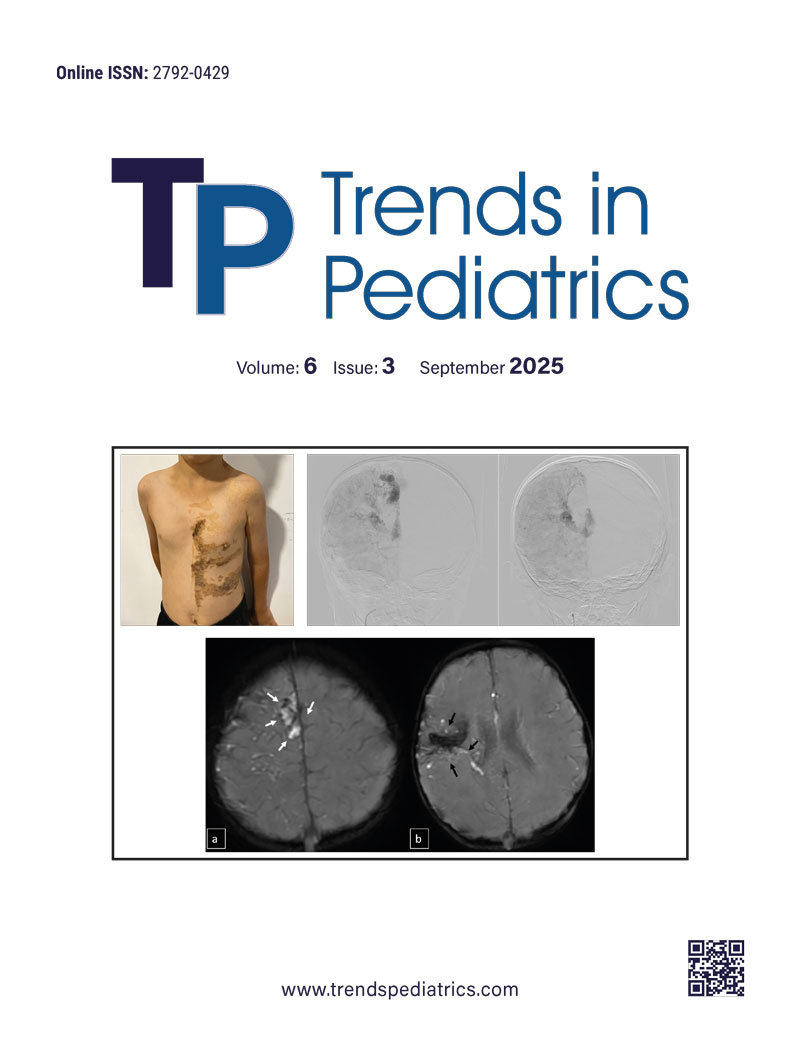
On physical examination, his height was 117 cm (-1.7 SDS), and his weight was 20 kg (-1.2 SDS). He had facial asymmetry and a hemorrhagic cavernous hemangioma on the right palatal mucosa. Additionally, a hyperpigmented, helical epidermal nevus was observed on the left side of the trunk, extending from the left shoulder to the lower abdomen and right gluteal region, following the ‘Blaschko’s lines’ along the left arm (Figure 1).
The patient’s initial work-up, including hematological, biochemical, thyroid functions, and coagulation parameters, was all within normal limits. Abdominal ultrasonography, cardiac echocardiography, skeletal survey, and ophthalmologic evaluation were also unremarkable. A pediatric genetics consultation was requested. Whole-exome sequencing (WES) revealed no pathogenic variants. Given the rarity and complexity of MMD, identifying a causative gene remains challenging. Since no specific genetic test is currently available for MMD, additional system evaluations such as ophthalmologic, cardiac, dermatologic, and skeletal assessments were recommended only in the presence of clinical suspicion. No associated genetic syndrome was identified upon re-evaluation by the genetics team.
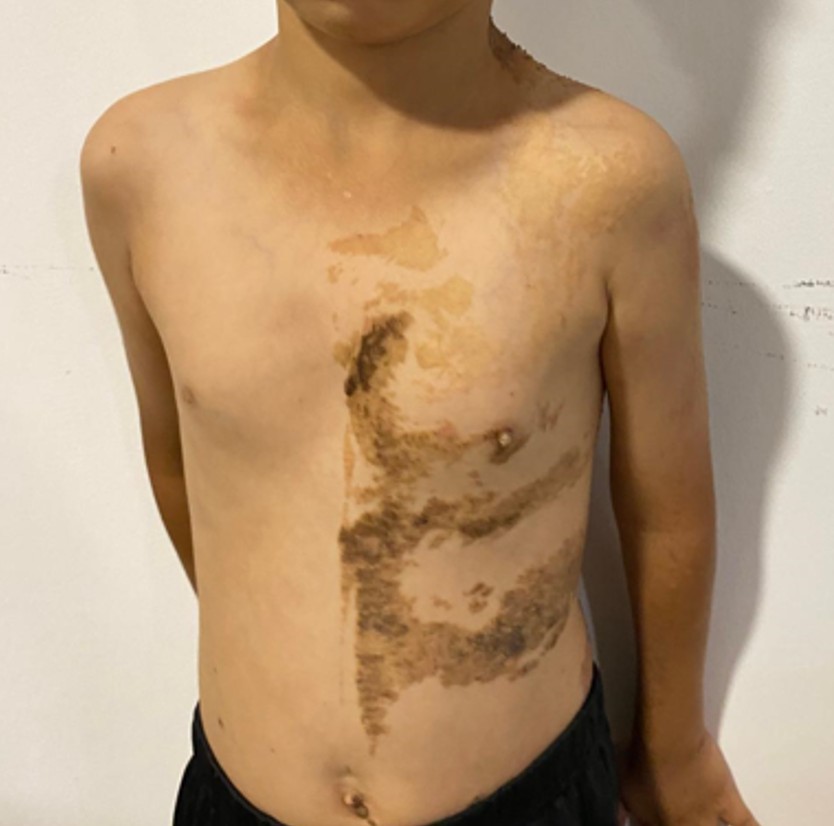
Cranial MRI demonstrated a dilated tortuous arteriovenous malformation in the right frontal lobe, fed by the distal branches of the anterior cerebral artery (ACA), draining into the cortical veins and central nervous system. A second AVM was detected in the corona radiata, with dilated parietotemporal draining veins fed by perisylvian branches of the middle cerebral artery (MCA), draining into the internal cerebral veins and the straight sinus via pericallosal dilated venous structures (Figure 2, Figure 3, Figure 4). At the level of the basal ganglia and mesencephalon, arterial angiomatosis resembling the classic “moyamoya” appearance was observed (Figure 5).
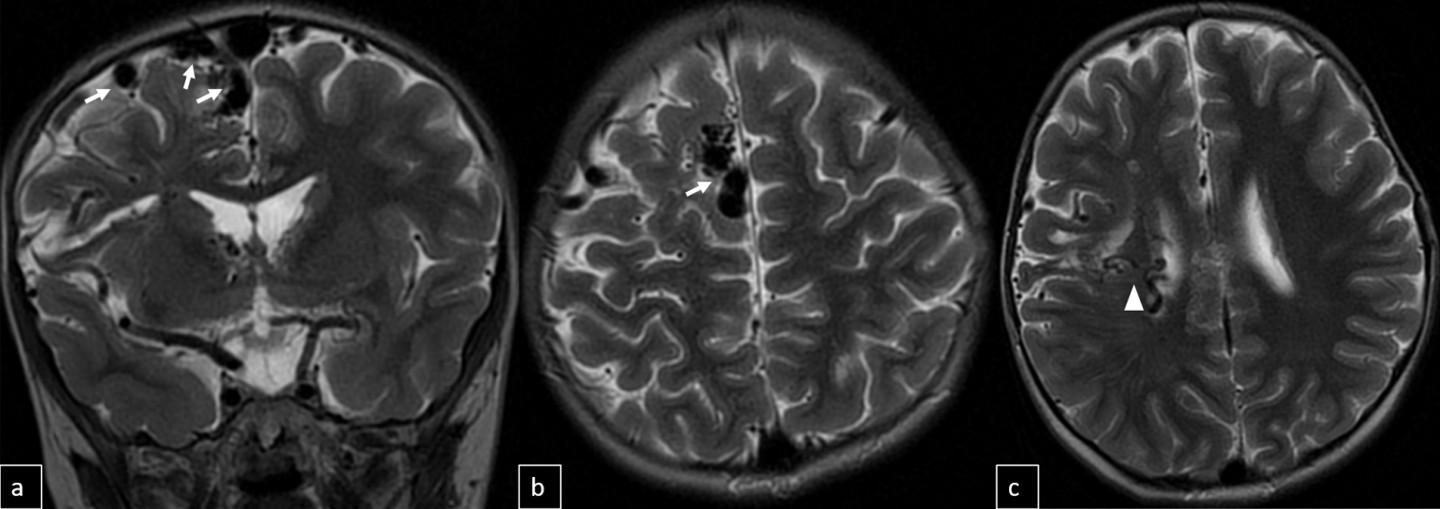
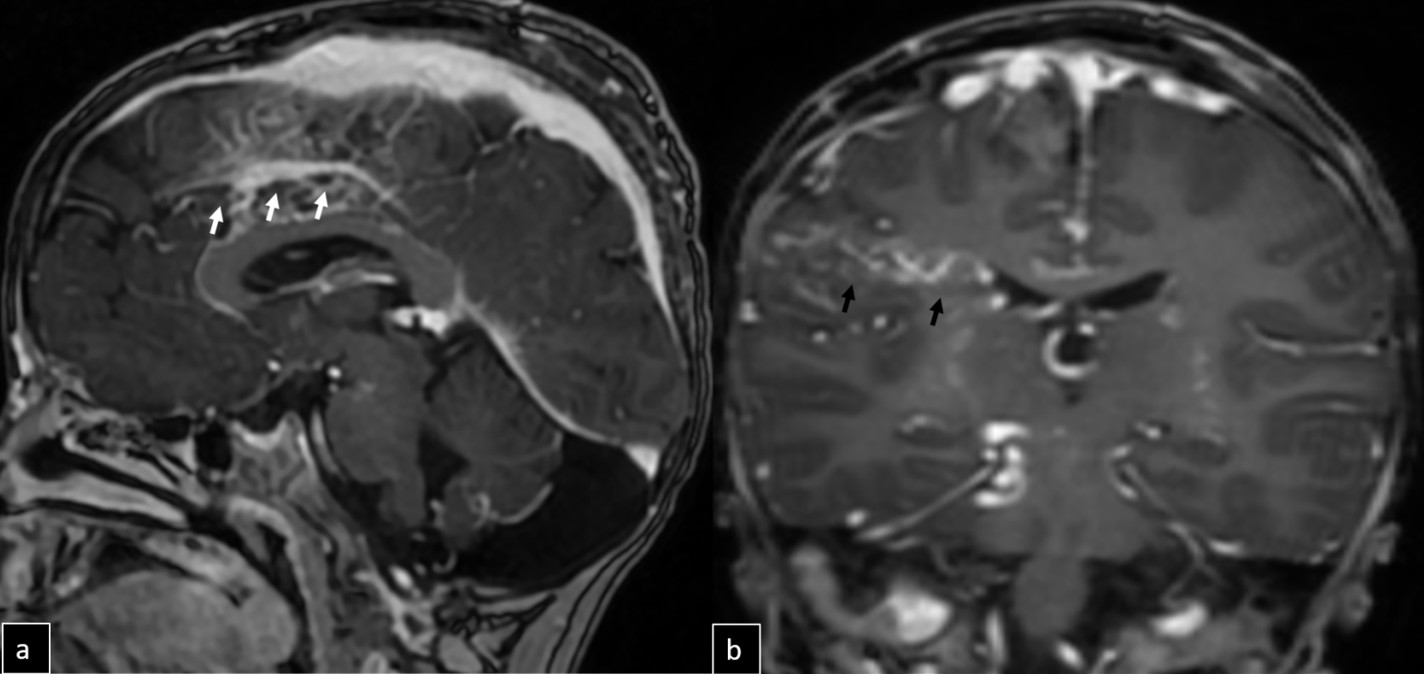
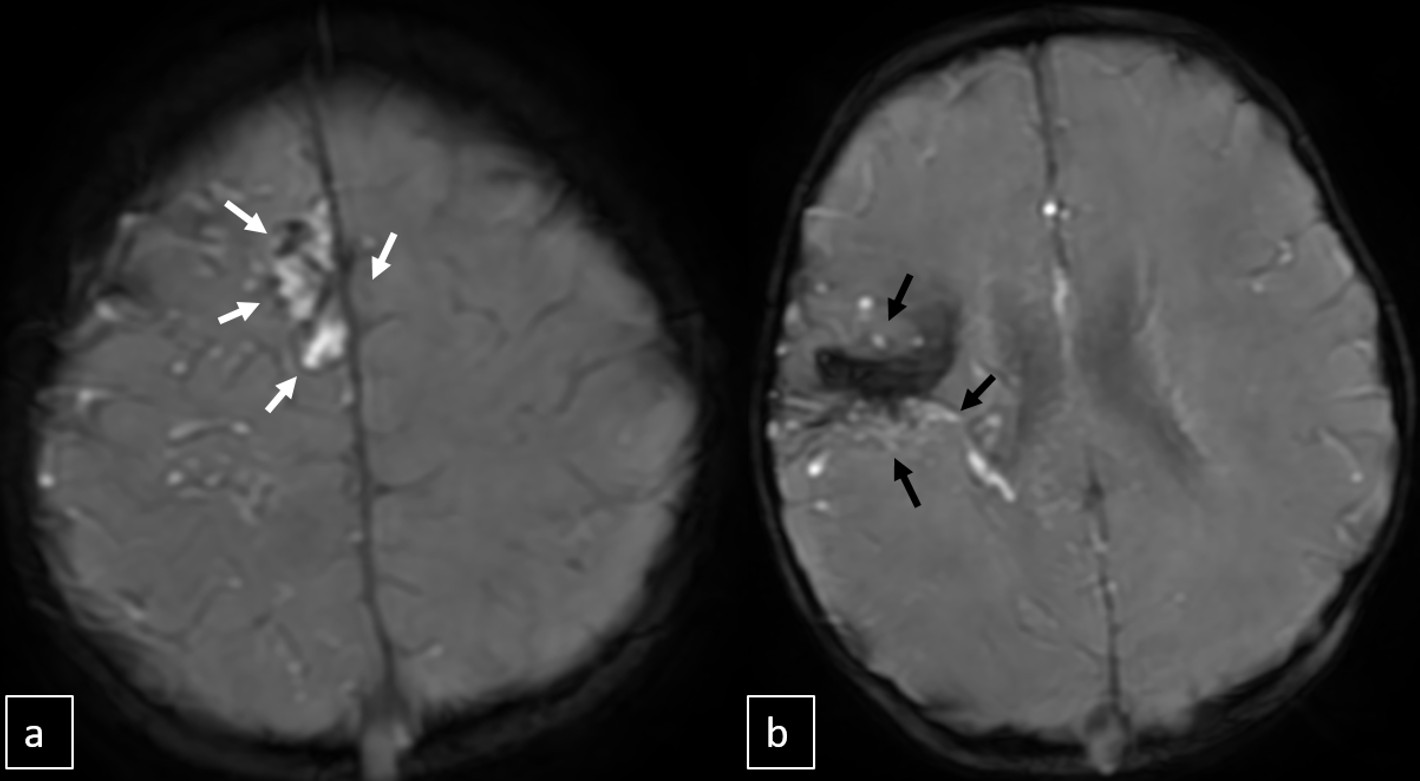
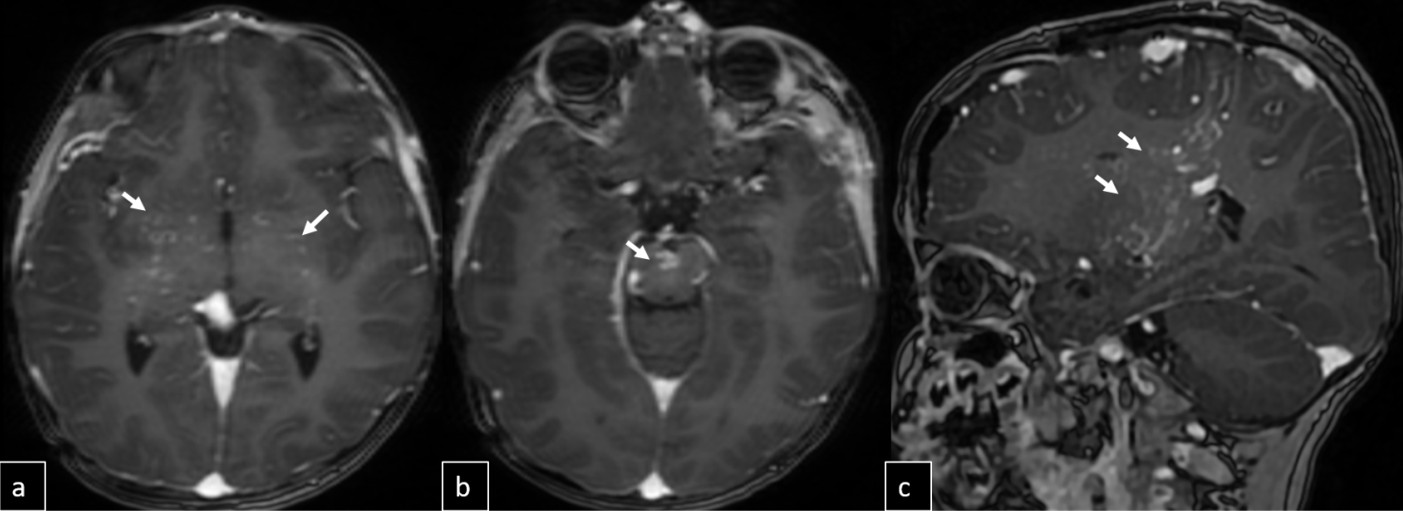
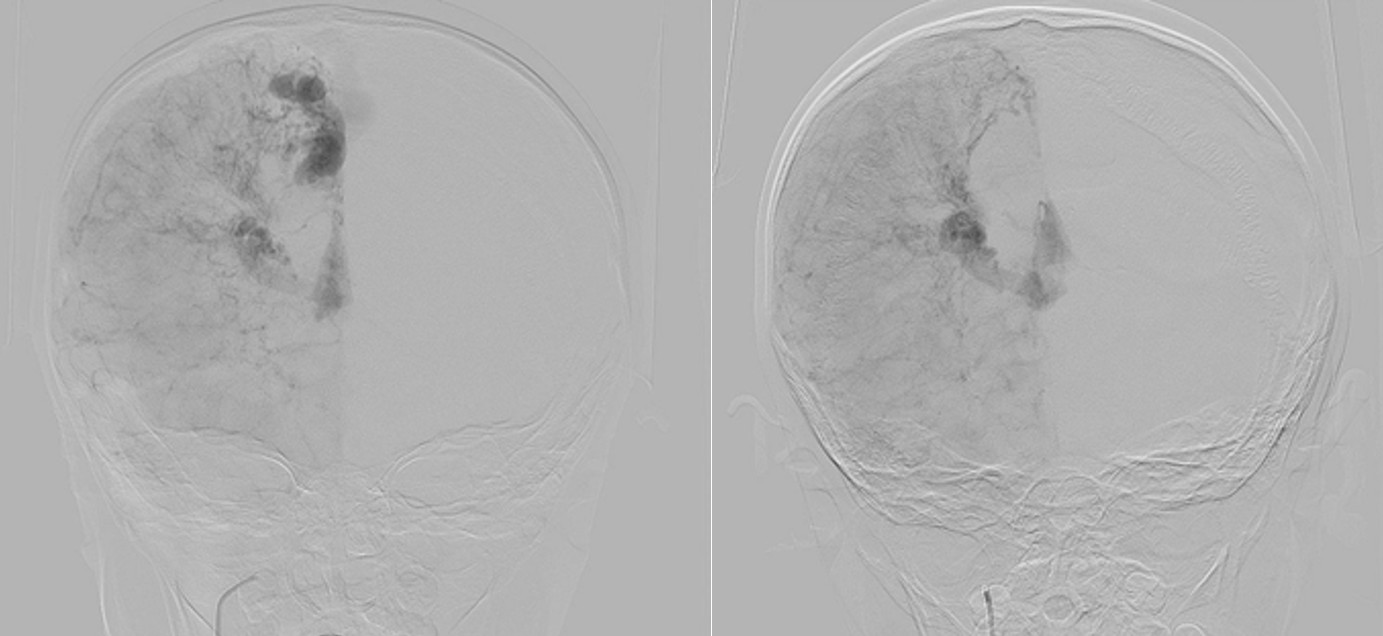
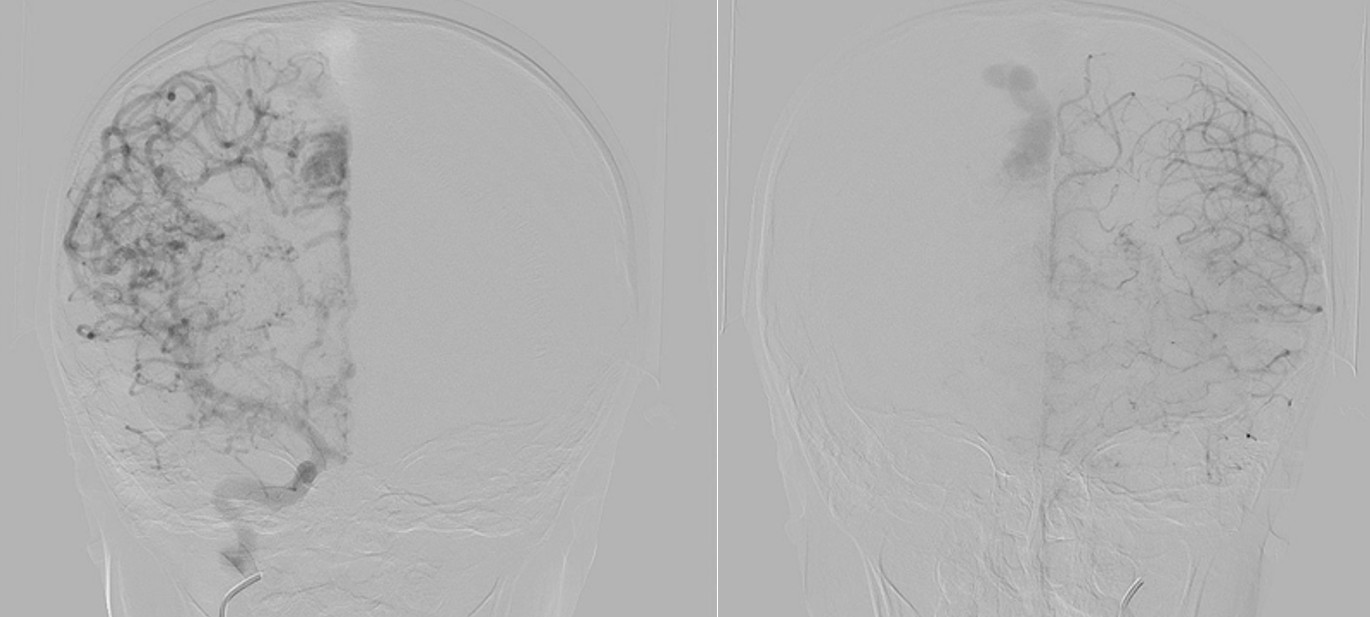
Digital subtraction angiography (DSA) confirmed the presence of two AVMs: one fed by distal branches of the right ACA and draining into the superior sagittal sinus, and another fed by MCA branches, draining into the internal cerebral vein. Capillary anastomoses were noted between the two AVMs. Selective bilateral internal carotid artery (ICA) injections revealed globally abnormal, tortuous arteriolar networks with delayed capillary filling, more pronounced on the right side (Figures 5, Figure 6, Figure 7). Based on these findings and the revised 2021 diagnostic criteria, the patient was diagnosed with MMD—characterized by stenosis of the intracranial ICA, narrowing at the terminal portion, and the development of abnormal collateral networks in the basal ganglia and periventricular white matter.4
The patient underwent surgical excision of the right parietal interhemispheric AVM. Postoperative follow-up showed no complications or neurological deficits, and no residual AVM was observed. Due to the proximity of the corona radiata AVM to the corticospinal tract, gamma knife radiosurgery was preferred and planned for a later age. Prophylactic low-dose aspirin was initiated to prevent ischemic events. Encephaloduroarteriosynangiosis (EDAS), an indirect cerebral revascularization technique, was considered for MMD, but was postponed due to potential bleeding risks in the presence of untreated AVMs. The patient continues to be followed up regularly without further complications.
DISCUSSION
MMD is a cerebrovascular disorder of unknown etiology. Although it can be associated with hereditary or systemic conditions, the underlying cause remains unidentified in more than half of pediatric patients.4-6 A genetic predisposition is suspected, particularly in patients with a positive family history.
Associated syndromes may present with a variety of systemic manifestations, including facial dimorphisms, cardiac anomalies, developmental delay, ophthalmologic abnormalities, short stature, macrocephaly, short neck, and cutaneous findings.3 In our patient, comprehensive clinical, radiological, laboratory, and multidisciplinary evaluations—including neurology, dermatology, and genetics—did not reveal any associated systemic disorder.
In children, MMD most frequently presents with ischemic rather than hemorrhagic strokes, with the former accounting for up to 80% of cases. Triggers such as crying, blowing, and hyperventilation can induce ischemic attacks through hypocapnia-induced vasoconstriction.7 Clinical manifestations may include headaches, seizures, and cognitive impairment. However, due to limited verbal communication skills in early childhood, diagnosis may be delayed.8 In our case, the diagnosis was made incidentally during the evaluation of intraoral bleeding from a cavernous hemangioma, prior to the onset of any ischemic or hemorrhagic neurological events.
Hemangiomas are benign vascular tumors resulting from vascular endothelial proliferation. Although rare, hemangiomas have been reported in association with MMD.3 In our patient, a hemorrhagic cavernous hemangioma was observed in the right palatal mucosa. Similar co-occurrences have been documented in the context of Morning Glory Disc Anomaly (MGDA) and Sturge-Weber syndrome.3 However, our patient did not exhibit any MGDA findings on fundoscopic examination, nor any facial port-wine stains, leptomeningeal angiomatosis, or glaucoma suggestive of Sturge-Weber syndrome.3 Dermatological findings reported in association with MMD include congenital melanocytic nevi, Becker nevi, and hairy pigmented macules.3 Our patient presented with a helical hyperpigmented epidermal nevus following Blaschko’s lines. Although the exact pathophysiological relationship between skin lesions and MMD remains unclear, some hypotheses suggest a shared origin in primary neuroectodermal dysgenesis or mesodermal anomalies during embryogenesis.9 Abnormal proliferation of smooth muscle cells, angiogenesis-related factors (e.g., fibroblast growth factors, hepatocyte growth factor), and adhesion molecules may contribute to both vascular malformations and cutaneous manifestations.10,11
The simultaneous presence of MMD and AVM is exceedingly rare, with few cases reported in the literature. It remains unclear whether this coexistence is coincidental or mechanistically linked. High-flow stress in AVMs may induce intimal hyperplasia in feeding arteries, leading to a moyamoya-like vascular response. Additionally, the abnormal angiogenic environment in MMD, characterized by increased expression of angiogenic and inflammatory mediators, may predispose to the formation of de novo AVMs or promote arteriovenous shunting through collateral pathways.12
DSA remains the gold standard for the diagnosis of MMD and associated vascular anomalies.4,13,14 Diagnosis is based on stenosis or occlusion of the distal internal carotid artery and/or vessels of the circle of Willis, accompanied by the development of prominent basal collateral networks.4,13,14 In our case, the diagnosis of MMD was confirmed through DSA following MRI findings.
Surgical revascularization is currently the most effective treatment modality in MMD.15 Antiplatelet agents are commonly used to reduce the risk of ischemic complications.15 However, treatment of MMD-associated AVMs remains controversial due to the delicate balance between preserving collateral circulation and preventing hemorrhage. Surgical resection of AVMs may risk disrupting collateral channels; thus, radiosurgical approaches such as gamma knife therapy are often preferred.12,15 In our case, gamma knife radiosurgery was scheduled for a later age due to the proximity of the AVM to the corticospinal tract. EDAS was considered but postponed due to bleeding risk in the presence of untreated AVMs.
The prognosis of MMD is influenced by patient age, severity of vascular involvement, presence of neurological symptoms, and coexisting vascular malformations.16 Our patient, who showed no neurological deficits and remains under close surveillance, exemplifies the importance of early diagnosis and individualized multidisciplinary management in rare pediatric cerebrovascular disorders.
This case illustrates a rare co-occurrence of MMD, cerebral AVM, and cutaneous lesions in a pediatric patient. The presence of atypical skin findings may provide early diagnostic clues for underlying neurovascular disorders. Timely recognition and a tailored multidisciplinary approach are essential to prevent complications and optimize long-term outcomes.
Ethical approval
This study has been approved by the Etlik City Hospital’s Ethics Committee (approval date 25.09.2024, number AEŞH-BADEK-2024-874). For case reports and for studies requiring informed consent, whether it was obtained should also be declared here.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Kim JS. Moyamoya disease: epidemiology, clinical features, and diagnosis. J Stroke. 2016;18:2-11. https://doi.org/10.5853/jos.2015.01627
- Smith ER, Scott RM. Moyamoya: epidemiology, presentation, and diagnosis. Neurosurg Clin N Am. 2010;21:543-51. https://doi.org/10.1016/j.nec.2010.03.007
- Mitri F, Bersano A, Hervé D, Kraemer M. Cutaneous manifestations in Moyamoya angiopathy: a review. Eur J Neurol. 2021;28:1784-93. https://doi.org/10.1111/ene.14754
- Kuroda S, Fujimura M, Takahashi J, et al. Diagnostic criteria for moyamoya disease - 2021 revised version. Neurol Med Chir (Tokyo). 2022;62:307-12. https://doi.org/10.2176/jns-nmc.2022-0072
- Goyal JP, Rao SS, Trivedi S. Moya moya disease in a child: a case report. Case Rep Neurol Med. 2011;2011:329738. https://doi.org/10.1155/2011/329738
- Bulut HT, Çoraplı M. Moyamoya disease in a pediatric case: A case report. J Surg Med. 2022;6:75-6. https://doi.org/10.28982/josam.1022512
- Nishimoto A, Ueta K, Onbe H. Cooperative study on Moyamoya disease in Japan. In: Abstracts of the 10th Meeting on Surgery for stroke. Tokyo: Nyuuron-sha; 1981: 53-8
- Williams TS, Westmacott R, Dlamini N, et al. Intellectual ability and executive function in pediatric moyamoya vasculopathy. Dev Med Child Neurol. 2012;54:30-7. https://doi.org/10.1111/j.1469-8749.2011.04144.x
- Sathyan S, Chackochan M. Morning glory disc anomaly and facial hemangiomas in a girl with moyamoya syndrome. Indian J Ophthalmol. 2018;66:1644-6. https://doi.org/10.4103/ijo.IJO_538_18
- Bersano A, Guey S, Bedini G, et al. Research progresses in understanding the pathophysiology of moyamoya disease. Cerebrovasc Dis. 2016;41:105-18. https://doi.org/10.1159/000442298
- Achrol AS, Guzman R, Lee M, Steinberg GK. Pathophysiology and genetic factors in moyamoya disease. Neurosurg Focus. 2009;26:E4. https://doi.org/10.3171/2009.1.FOCUS08302
- Noh JH, Yeon JY, Park JH, Shin HJ. Cerebral arteriovenous malformation associated with moyamoya disease. J Korean Neurosurg Soc. 2014;56:356-60. https://doi.org/10.3340/jkns.2014.56.4.356
- Smith JL. Understanding and treating moyamoya disease in children. Neurosurg Focus. 2009;26:E4. https://doi.org/10.3171/2000.01.FOCUS08306
- Sfaihi L, Elloumi S, Fourati H, Kamoun T, Mnif Z, Hachicha M. Arterial ischemic stroke in children: 22 cases from southern Tunisia. Fetal Pediatr Pathol. 2013;32:271-5. https://doi.org/10.3109/15513815.2012.754523
- Arias EJ, Derdeyn CP, Dacey RG, Zipfel GJ. Advances and surgical considerations in the treatment of moyamoya disease. Neurosurgery. 2014;74(Suppl 1):S116-25. https://doi.org/10.1227/NEU.0000000000000229
- Pandey P, Steinberg GK. Neurosurgical advances in the treatment of moyamoya disease. Stroke. 2011;42:3304-10. https://doi.org/10.1161/STROKEAHA.110.598565
Copyright and license
Copyright © 2025 The author(s). This is an open-access article published by Aydın Pediatric Society under the terms of the Creative Commons Attribution License (CC BY) which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.