Abstract
Type 1 interferonopathies are rare genetic disorders characterized by abnormal type 1 interferon (IFN) signaling. They cause chronic inflammation and multisystemic symptoms typically present in early childhood. Neurological, dermatological, and musculoskeletal features are common and often resistant to conventional therapies. Mutations in genes involved in nucleic acid sensing, degradation, proteasome function, and IFN signaling lead to the accumulation of self-deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) in the cytoplasm and a sustained type 1 IFN response, causing tissue damage and autoimmunity. This group includes various syndromes, such as Aicardi-Goutières syndrome (AGS), STING-associated vasculopathy (SAVI), and COPA syndrome. Diagnosis involves clinical evaluation, IFN signature analysis, and genetic testing. Treatment aims to modulate the IFN response by using JAK inhibitors, anti-IFN antibodies, and reverse transcriptase inhibitors. However, these therapies are not curative and have limited efficacy. Further research is needed to develop targeted treatments and improve outcomes, and a multidisciplinary management approach is essential because of the complexity and rarity of these disorders.
Keywords: Aicardi-Goutières syndrome, anifrolumab, baricitinib, interferons, Janus kinases, sifalimumab
INTRODUCTION
Type 1 interferonopathies are a new group of rare and severe multisystemic disorders characterized by abnormal and uncontrolled activation of type 1 interferon (IFNs) signaling pathways caused by specific genetic mutations in the immune system.1,2 The basic mechanism underlying these diseases is that the immune system mistakenly recognizes its own deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) as a pathogen.3 This leads to chronic inflammation, resulting in persistently high levels of cytokines, especially IFN-α and IFN-β, and the emergence of autoimmune and autoinflammatory responses.1,4
IFNs play a key role in the immune system’s defense against viral, bacterial, and tumoral pathogens. Type I IFNs (particularly IFN-α and IFN-β) are produced by almost every nucleated cell, forming the first line of defense in innate immunity and providing a rapid antiviral response.1 These increase pathogen resistance by regulating cellular functions. Type II IFNs are composed solely of IFN-γ and are secreted by T cells and natural killer cells, supporting adaptive immunity.3 IFN-γ activates macrophages and potentiates the cytotoxic T-cell response. Type III IFNs (IFN-λ) act at the entry points of infection and induce a localized antiviral response in the respiratory and digestive epithelia.4 Although IFNs have positive effects on the human body, prolonged IFN exposure in animal models has been linked to growth retardation and organ damage. Interferonopathies often affect the central nervous system, skin, and joint tissues, presenting symptoms from neurological dysfunction to skin lesions.1,3
Type 1 interferonopathies typically begin in early childhood, but the onset age varies depending on the specific disease and genetic mutations.5 Aicardi-Goutières syndrome (AGS), the first identified Mendelian type I interferonopathy, is a multisystem disorder with neurological symptoms resembling congenital viral infections.1,4 In AGS, symptoms usually appear in infancy, whereas other interferonopathies may not show symptoms until childhood or, rarely, adolescence.3
In recent years, interferonopathies have taken place in modern medicine as rare genetic diseases due to reports of similar cases and an understanding of new genetic mutations and disease mechanisms.3 This review aims to provide an overview of this rare and current disease group.
PATHOGENESIS
Type 1 interferonopathies occur when endogenous nucleic acids accumulate in the cytoplasm, usually as a result of cellular stress, infections, or genetic mutations.1 In this process, intracellular DNA and RNA in particular activate cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS) and stimulator of interferon genes (STING) molecules, which act as nucleic acid sensors, leading to excessive production of type 1 IFN.6 While the cGAS-STING pathway initiates the immune system’s natural response to infections and cellular damage, uncontrolled activation of this mechanism leads to chronic production of type 1 IFNs and perpetuation of the inflammatory response.6 Furthermore, cGAS sensing of mitochondrial DNA and other nucleic acids released from damaged cells promotes sustained activation of the interferon response through STING.1 Defects in the proteasome and autophagy systems further exacerbate the pathogenesis by increasing the accumulation of these nucleic acids in the cytoplasm. We tried to summarize the overall process of the type I IFN response in Figure 1.
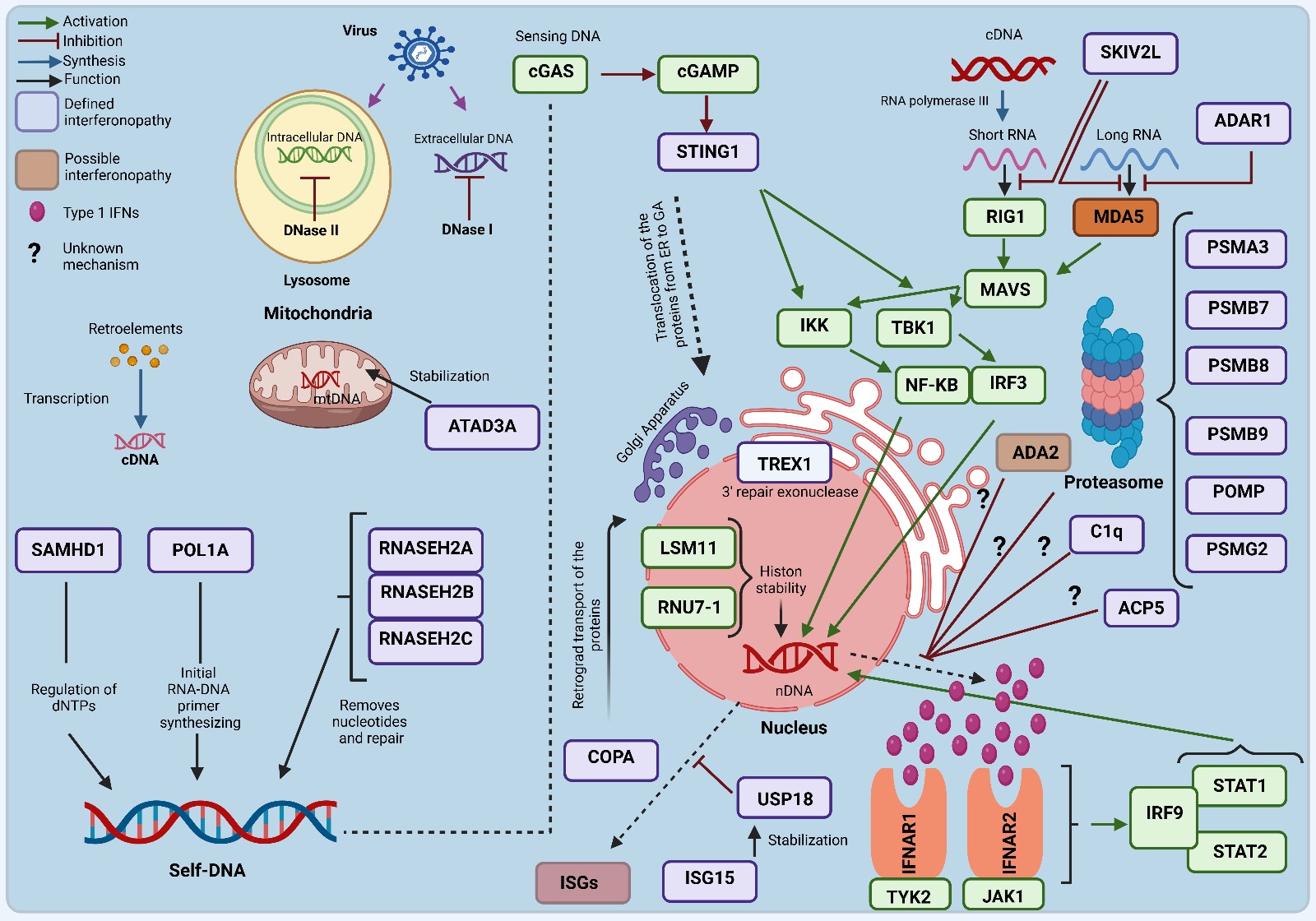
(ACP5: Acid Phosphatase 5, Tartrate-Resistant, ADA2: Adenosine Deaminase 2, ADAR1: Adenosine Deaminase Acting on RNA 1, ATAD3A: ATPase Family AAA Domain-Containing Protein 3A, C1q: Complement Component 1q, cGAS: cyclic guanosine monophosphate-adenosine monophosphate synthase, COPA: Coatomer Protein Complex Subunit Alpha, DNA: Deoxyribonucleic Acid, DNase I: deoxyribonuclease I, DNase II: deoxyribonuclease II, dNTP: Deoxynucleotide triphosphate, IFNAR1: Interferon Alpha/Beta Receptor 1, IFNAR2: Interferon Alpha/Beta Receptor 2, IRF3: Interferon Regulatory Factor 3, IRF9: Interferon Regulatory Factor 9, JAK1: Janus Kinase 1, LSM11: LSM11, U7 Small Nuclear RNA Associated, MAVS: Mitochondrial antiviral signaling protein, MDA5: Melanoma differentiation-associated protein, mtDNA: Mitochondrial DNA, NF-κB: Nuclear factor κB, POL1A: DNA Polymerase Alpha 1, POMP: Proteasome Maturation Protein, PSMA3: Proteasome Subunit Alpha Type-3, PSMB7: Proteasome Subunit Beta Type-7, PSMB8: Proteasome Subunit Beta Type-8, PSMB9: Proteasome Subunit Beta Type-9, PSMG2: Proteasome Assembly Chaperone 2, RNA: Ribonucleic Acid, RNASEH2A: Ribonuclease H2 Subunit A, RNASEH2B: Ribonuclease H2 Subunit B, RNASEH2C: Ribonuclease H2 Subunit C, RIG1: Retinoic acid-inducible gene-1, RNU7-1: RNA, U7 Small Nuclear 1, SAMHD1: Sterile Alpha Motif, and Histidine-Aspartate Domain-Containing Protein 1, SKIV2L: Ski2 Like RNA Helicase, STAT1: Signal Transducer and Activator of Transcription 1, STAT2: Signal Transducer and Activator of Transcription 1, STING: stimulator of interferon genes, IKK: Inhibitor of κB Kinase, TBK1: TANK-binding kinase 1, IFN: Interferon, TREX1: Three Prime Repair Exonuclease 1, TYK2: Tyrosine Kinase 2, USP18: Ubiquitin-Specific Peptidase 18).
Deoxyribonucleic Acid (DNA) sensing
Normally, most DNA resides in the cell nucleus. The immune system interprets DNA in the cytoplasm as a potential viral or bacterial infection, signaling danger.7 Type 1 interferonopathies are disorders characterized by excessive activation of the immune system, resulting from viral infections or genetic mutations that cause cellular misidentification of endogenous DNA, termed “self-DNA,” as exogenous material.7 Under normal physiological conditions, exogenous DNA within the cellular environment is eliminated by enzymatic processes, primarily through the action of cytosolic deoxyribonuclease I (DNase I) and DNase II. However, in case of loss of function of enzymes such as Three Prime Repair Exonuclease 1 (TREX1), a cytosolic DNase located in the nuclear membrane, the cell accumulates its own DNA or damaged DNA.8
Proper processing of intracellular DNA is ensured by proteins such as DNA Polymerase Alpha 1 (POL1A), Sterile Alpha Motif, and Histidine-Aspartate Domain-Containing Protein 1 (SAMHD1), as well as ribonucleotide scavenger enzymes such as Ribonuclease H2 (RNASEH2) A, RNASEH2B, and RNASEH2C.5 SAMHD1 regulates deoxynucleotide triphosphate (dNTP) levels, keeping DNA synthesis under control and preventing self-DNA accumulation. However, LSM11, U7 Small Nuclear RNA Associated (LSM11) and RNA, U7 Small Nuclear 1(RNU7-1) mutations resulting from defects in histone pre-mRNA processing can lead to leakage of this DNA into the cytosol.9 ATPase Family AAA Domain-Containing Protein 3A (ATAD3A) mutations can also lead to disruption of the integrity of the mitochondrial membrane and escape of mitochondrial DNA into the cytoplasm.10
DNA passing into the cytosol is sensed by the enzyme cGAS, and this sensing initiates the production of the molecule cGAMP, which activates the STING protein in the endoplasmic reticulum (ER).11,12 The STING protein is located on intracellular membranes and is a critical signaling pathway for initiating the immune response.13 Mutations in genes such as coatomer subunit alpha (COPA), Transmembrane Protein 173 (TMEM173), and TREX1 lead to abnormal activation of the STING signaling pathway, resulting in sustained and uncontrolled production of type 1 IFN.14
Ribonucleic Acid (RNA) sensing
Detection of viral RNA in the cytoplasm signals danger to the immune system and triggers an innate immune response. This process involves two main RNA sensors: retinoic acid-inducible gene-1 (RIG-1) and melanoma differentiation-associated protein (MDA5). Both belong to the RIG-1-like receptor (RLR) family and initiate IFN production by recognizing viral RNA.15,16 RIG-1 (encoded by DDX58) detects short double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA) molecules with triphosphate groups at their ends, whereas MDA5 (encoded by IFIH-1) binds to longer dsRNA molecules. When both sensors are activated, they transmit the signal to the mitochondrial antiviral signaling protein (MAVS) adaptor protein located in the cytosol.16,17 MAVS is located on the mitochondrial outer membrane and interacts with TNF receptor-associated factors (TRAF) proteins and activates downstream kinases.18
Common pathway in nucleic acid sensing
Stimulation of the STING pathway by DNA or the MAVS pathway by RNA initiates immune signaling. STING activity is inhibited by enzymes such as cGAMP phosphodiesterase (cGAMP-PDE), while the MAVS signaling pathway is repressed by RNF125 ubiquitin ligase.19 A defect in the function of these regulatory proteins leads to over-activation of the STING and MAVS signaling pathways, activating the TANK-binding kinase 1 (TBK1) inhibitor and IKKε kinase complex. TBK1 activates IFN regulatory factor 3 (IRF3). Activation of IKKε leads to the activation of the nuclear factor κB (NF-κB) transcription factor.20
Proteasomes in this context
Proteasomes maintain intracellular protein balance, clear misfolded proteins, and support immune responses by promoting antigen presentation.21 Mutations in proteasome-related genes such as PSMA3, PSMB7, PSMB8, PSMB9, POMP, and PSMG2 disrupt the function of the proteasome complex, leading to an uncontrolled type 1 IFN response.22 This dysfunction causes ER accumulation of misfolded proteins and activates the ER membrane protein Inositol-Requiring Enzyme 1 (IRE1). Activated IRE1 stimulates transcription factors such as IRF3 and NF-κB, triggering the production of proinflammatory cytokines and IFNs.23
Nuclear response
The nuclear response begins with the recognition of cytosolic DNA or RNA in immune cells. It proceeds when transcription factors are phosphorylated and transported to the nucleus to initiate the expression of type 1 IFN and proinflammatory cytokines.24 Factors such as IRF3, IFN regulatory factor 7 (IRF7), and NF-κB play key roles in this process. The cGAS-STING pathway activates IRF3 in response to DNA, while the MAVS pathway activates IRF7 and NF-κB in response to RNA.12 IRF3 is phosphorylated and transported to the nucleus via TBK1 kinase to initiate the transcription of type 1 IFN and interferon-stimulated gene (ISG). NF-κB enhances inflammatory cytokine production, leading to migration of immune cells to the site of infection.25 Furthermore, Signal Transducer and Activator of Transcription (STAT) 1 and STAT2 sustain the antiviral response by activating the Janus kinase (JAK) - STAT pathway and potentiate the expression of antiviral proteins such as Interferon-Stimulated Gene 15 (ISG15) and Myxovirus Resistance Protein 1 (MX1). Since excessive nuclear response can trigger autoimmune responses, proteins such as Suppressor of Cytokine Signaling (SOCS) 1 and SOCS3 limit the response by inhibiting the JAK-STAT pathway; Protein Inhibitor of Activated STAT (PIAS) 1 and PIAS3 control STAT1 and STAT3 activity.26 Ubiquitin-Specific Peptidase 18 (USP18), stabilized by ISG15, plays a negative regulatory role in ISG transcription, optimizing the antibacterial response against mycobacteria.27 Osteopontin (OPN) promotes type I IFN production, whereas this effect is limited by tartrate-resistant acid phosphatase (ACP5).28 Complement proteins such as C1q also contribute to immune balance by inhibiting ISG transcription.
Type I IFN activity
During infection, recognition of viral RNA and DNA in the cell cytoplasm initiates type I IFN production via RLRs and cGAS-STING, activating transcription factors such as IRF3 and NF-κB.29 This, in turn, leads to the production of IFN-α and IFN-β, resulting in the secretion of these molecules. IFN-α and IFN-β bind to IFN-alpha/beta receptors (IFNAR) consisting of IFNAR1 and IFNAR2 subunits on the cell surface by autocrine or paracrine action.30 This binding activates the JAK-STAT pathway; IFNAR-bound Tyrosine Kinase 2 (TYK2) and JAK1 kinases are activated through phosphorylation and subsequent phosphorylation of STAT1 and STAT2 proteins.31 Phosphorylated STAT1 and STAT2 combine with IRF9 to form the Interferon-Stimulated Gene Factor 3 (ISGF3) complex. The ISGF3 complex then enters the nucleus and initiates ISG transcription. ISGs encode proteins that inhibit viral replication and exert antiviral effects. Antiviral proteins, such as Protein Kinase R (PKR), 2’-5’-Oligoadenylate Synthetase (OAS), and Myxovirus Resistance Protein A (MxA), stop virus replication in infected cells, limiting the spread of infection. Thus, the type I IFN signaling pathway provides the immune system’s initial response by creating a strong antiviral environment in both infected cells and surrounding cells.32 Negative regulatory proteins such as SOCS1 and SOCS3 fail to inhibit the JAK-STAT pathway, and proteins such as USP18 fail to suppress the sustained activation of IFNAR, resulting in prolonged type I IFN activation and tissue damage.33
CLINICAL FEATURES
Type 1 interferonopathies are autoinflammatory and autoimmune disorders characterized by onset in early childhood due to genetic alterations. Neurological manifestations include microcephaly, seizures, spasticity, and intracranial calcification, while dermatological findings encompass livedo reticularis, lupus-like rashes, and ulceration. Additional common features include retinal vasculopathy, acrocyanosis, finger necrosis, interstitial lung diseases (ILD), bone marrow depression, growth retardation, hepatosplenomegaly, and arthritis.1,5 The general genetic and clinical features of these diseases are summarized in Table 1.
| AD: autosomal dominant; AGS: Aicardi-Goutières syndrome; AR: autosomal recessive; DADA2: adenosine deaminase 2 deficiency; FCL: familial chilblain lupus; FSLE: familial systemic lupus erythematosus; HSM: hepatosplenomegaly; ISG15: interferon-stimulated gene 15 deficiency; PRAAS: proteasome-associated autoinflammatory syndrome; SAVI: STING-associated vasculopathy with onset in infancy; SLE: systemic lupus erythematosus; SMS: singleton–Merten syndrome; SPENCD: spondyloenchondrodysplasia; THES: trichohepatoenteric syndrome; USP18: ubiquitin-specific protease 18 deficiency; RVCL: retinal vasculopathy with cerebral leukodystrophy; XLRPD: X-linked reticulate pigmentary disorder. | |||
| Table 1. Summary of Inheritance, Genetic, and Clinical Characteristics of Type I Interferonopathies | |||
| Inheritance | Gene | Clinical Features | |
| AGS | AR/AD | SAMHD1, ADAR1, RNASEH2A, RNASEH2B, RNASEH2C, TREX1, IFIH1 | mental-motor retardation, developmental delay, spasticity, dystonia, ataxia, peripheral neuropathy, epilepsy, brain atrophy (cortical and subcortical), leukodystrophy, intracranial calcification, anemia, thrombocytopenia, chilblain, livedo, HSM |
| COPA | AD | COPA | peripheral arthritis, interstitial lung disease, diffuse alveolar hemorrhage, glomerulonephritis, oral ulcer, livedo |
| DADA2 | AR | ADA2/CECR1 | Growth retardation, recurrent fever, livedo, digital gangrene, stroke, epilepsy, peripheral neuropathy, pancytopenia, hemolytic anemia, mesenteric ischemia |
| FCL | AD | TREX1, SAMHD1 | Early-onset, cold-induced chilblains in acral sites, arthralgia, lymphopenia, myalgia, conjunctivitis |
| FSLE | AR/AD | TREX1, SAMHD1, ACP5, DNASE1, DNASE1L3, PRKCD, C1Q/R/S, C2, C3, C4 | Arthritis, lupus nephritis, malar rash, discoid rash, photosensitivity, chilblain, pancytopenia, pericarditis, myocarditis, intracranial calcification, leukodystrophy, vision loss, stroke, seizure, psychosis |
| ISG15 | AR | ISG15 | increased susceptibility to bacterial and viral infections, mycobacterial infections, seizures, developmental delay, muscle weakness, skin rash |
| PRAAS | AR | PSMA3, PSMB7, PSMB8, PSMB9, POMP, PSMG2 | recurrent episodes of fever from infancy, neutrophilic dermatosis, lipodystrophy, bone tenderness, muscle weakness |
| SAVI | AD | TMEM173/STING1 | cold-induced livedo, ulceration, necrosis of fingertips, short fingers, interstitial lung disease, arthritis, fever, myalgia |
| SMS | AD | IFIH1, DDX58 | early tooth loss starting in childhood, enamel hypoplasia, skeletal dysplasia, deformity of the hands and feet, calcification and bone hardening, vascular calcification |
| SPENCD | AR | ACP5 | developmental defects in the spine (spondylo-) and long bones (endochondro-), growth retardation, short stature, muscle weakness, spasticity, autoimmune thyroid diseases |
| THES | AR | SKIV2L, TTC37 | severe and treatment-resistant diarrhea, malabsorption, growth retardation, hepatosplenomegaly, cirrhosis, trichorexis nodosa, dry skin, skin hyperpigmentation, eczematous rash, pancytopenia |
| USP18 | AD | USP18 | severe skin rashes, fever, muscle weakness, respiratory distress, seizures, starting in the neonatal period |
| RVCL | AD | TREX1 | retinal vasculopathy, cerebral leukoencephalopathy, hypertension, renal failure, difficulty speaking and walking |
| XLRPD | AR | POLA1 | reticular hyperpigmentation, photophobia, corneal opacity, recurrent pneumonia, nephritis, renal failure |
Aicardi-Goutières Syndrome (AGS)
AGS is a rare autoinflammatory disease in patients with type 1 interferonopathies and is characterized by neurological symptoms in early childhood. Mutations in genes such as TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1 lead to the accumulation of DNA or RNA in the cytoplasm, triggering the continuous production of type 1 IFN and causing the body to attack its own tissues.1,5,34
In the neonatal period, AGS presents clinical features resembling congenital infections, including TORCH. During this period, cerebral calcifications, particularly in the basal ganglia and other cerebral regions, are often accompanied by microcephaly and are crucial for diagnosing the condition. Cerebral white matter damage and encephalopathy symptoms resulting from elevated type 1 IFN levels are also characteristic findings. Furthermore, hepatosplenomegaly, increased leukocyte levels, and febrile episodes frequently accompany the disease presentation.5
The most prominent findings that shape the clinical picture of AGS in the infantile period include developmental delay, spasticity, increased muscle tone, and seizures. In addition to severe motor and mental retardation, significant neurologic symptoms, such as limitation of limb movements and inadequate head control, are observed in children during this period. Magnetic resonance imaging (MRI) findings reveal basal ganglia calcifications and white matter damage.5 In addition, livedo reticularis, lace-like bruises on the skin such as acrocyanosis and hair loss, are among the autoinflammatory manifestations specific to the infantile phase of AGS.4
In late childhood, AGS is characterized by marked autoimmune and neurological disorders, as well as exacerbated autoinflammatory symptoms. Later in life, sustained immune system activation may cause autoimmune responses such as lupus-like skin lesions, lace-like rashes, and arthritis. Some patients develop permanent neurological damage, such as intellectual disability, vision, and hearing loss.1,5 Especially in those with IFIH1 mutation, autoimmune findings are more prominent, and clinical features such as skin atrophy and hair loss are frequently seen during this period.1
STING-Associated Vasculopathy with Onset in Infancy (SAVI)
Gain-of-function (GOF) mutations in STING1, which encodes a protein called STING, underlie the disease. These mutations lead to sustained activation of STING and consequent chronic stimulation of type 1 IFN production.35 SAVI causes severe inflammation of the skin, lungs, and vascular system, usually starting in childhood, and can lead to permanent tissue damage.36
The most characteristic findings of SAVI include livedo reticularis, ulcerations, and necrosis of the fingertips, which are particularly severe in cold weather. These findings usually begin shortly after birth and can lead to tissue loss, shortening, and deformation of the fingers.37 A large proportion of patients develop ILD, which can lead to severe respiratory failure and a marked decrease in quality of life. Early onset of pulmonary involvement is a factor that adversely affects the prognosis of the disease; therefore, close monitoring of pulmonary findings is important.37 Fever, musculoskeletal pain, arthritis, anorexia, growth, and developmental delay may also be observed.35,36 Rare cases of alopecia, short stature, epilepsy, intracranial calcification, pulmonary hypertension, and spastic diplegia have also been reported.37
COPA syndrome
Heterozygous mutations in the COPA gene constitute the basic mechanism of the disease by disrupting the function of the COPA protein, which regulates intracellular protein transport and the return of proteins between the endoplasmic reticulum and the Golgi body. This dysfunction leads to intracellular accumulation of misfolded or mutated proteins, resulting in increased production of type 1 IFN.38
COPA syndrome is one of the most common diseases with lung involvement and is characterized by inflammatory conditions, particularly ILD, and alveolitis. These patients usually present with symptoms such as shortness of breath, cough, and breathing difficulties, which usually begin at an early age.39 It is characterized by joint involvement, usually presenting as symmetrical polyarthritis and morning stiffness. In some cases, signs of vasculitis, such as lupus-like skin rashes, livedo reticularis, and ulceration, have also been reported.38 Fever, growth retardation, and glomerulonephritis may also be seen.38
Familial Systemic Lupus Erythematosus (SLE)
Familial SLE is an autoimmune disease that affects multiple family members owing to a genetic predisposition, with early onset and severe progression. Key mutations increasing SLE susceptibility are found in TREX1, DNASE1L3, BLK, IRAK1, SAMHD1, ACP5, DNase1, protein kinase C δ (PRKCD), and early complement proteins (C1q/r/s, C2, C3, and C4).40,41 Specifically, TREX1 and DNASE1L3 mutations lead to DNA accumulation in the cytosol, stimulating type 1 IFN production and causing skin, joint, and kidney damage.42 Skin manifestations include malar rash and chilblain lesions, worsened by sun exposure.40 Joint symptoms include symmetrical pain, morning stiffness, and arthritis, impacting quality of life despite not causing permanent deformity. Lupus nephritis, which is common in early-onset familial SLE, can result in permanent kidney damage.42 Rarely, retinal vasculopathy, leukodystrophy, vision loss, stroke, and mental retardation may occur. It is highly resistant to standard SLE treatments due to the constant activation of the immune system.42
Familial Chilblain Lupus (FCL)
FCL is caused by heterozygous mutations in TREX1 or SAMHD1, resulting in intracellular DNA accumulation.1,5 The hallmark of FCL is red-purple skin lesions on the fingertips, hands, feet, nose, and ears, which typically intensify in cold weather. These painful lesions can result in ulceration, necrosis, and tissue loss after prolonged exposure to cold.8 Phenotypic diversity should be indicated in FCL patients since patients with the same mutations may result in different severities of clinical features.43 Arthralgia and myalgia may also be seen but are usually less severe than skin symptoms. Lymphopenia and anti-nuclear antibody (ANA) positivity have been observed in some cases.1
Spondyloenchondrodysplasia (SPENCD)
SPENCD is a clinical entity characterized by autoimmune disorders and skeletal dysplasia caused by mutations in ACP5. This gene encodes tartrate-resistant acid phosphatase (TRAP), which is found in osteoclasts and is crucial for bone metabolism and immunity. Reduced TRAP activity impairs osteoclast function, affecting bone growth and the immune response.9,44 Short bones, metaphyseal dysplasia, platyspondyly, and developmental defects of the spine and long bones can be seen and can cause growth retardation and short stature.44,45 The disease is often linked to autoimmune conditions such as autoimmune thyroid diseases, SLE, Sjögren’s syndrome, and vasculitis. Headaches, spasticity, muscle weakness, and motor disorders may also occur.45
Deficiency of Adenosine Deaminase 2 (DADA2)
The literature does not definitively classify DADA2 as a type 1 interferonopathy, but it is believed to trigger a type 1 IFN response via STING, sustaining the immune response.46 DADA2 is an autosomal recessive (AR) inherited immunodeficiency syndrome caused by mutations in the ADA2 or CECR1 gene.9
One of the most characteristic features of DADA2 is the presence of vasculitis affecting small- and medium-sized vessels. This can lead to the development of livedo reticularis of the skin, ulceration, and gangrene of the fingers and toes. If vascular inflammation involves brain vessels, recurrent episodes of ischemic stroke may occur. Stroke, especially in childhood, is one of the remarkable clinical manifestations of DADA2.47
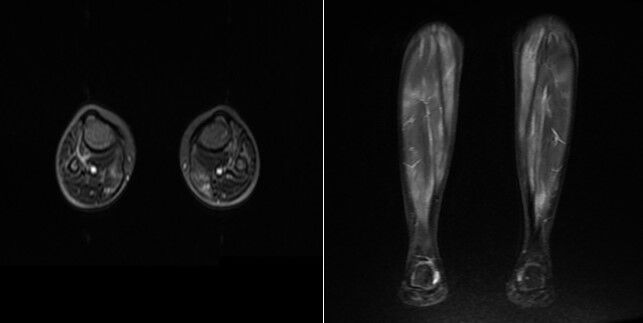
The inflammatory response triggered by ADA2 deficiency can lead to the development of hematologic abnormalities such as autoimmune hemolytic anemia, thrombocytopenia, and neutropenia. Systemic manifestations such as pancreatitis, hepatosplenomegaly, and arthritis are also included in the clinical spectrum of the disease.48 MRI findings of our patient with DADA2 are shown in Figure 2.
Trichohepatoenteric Syndrome (THES)
THES is a rare AR disease that occurs in early childhood and affects multiple organs, including the digestive system, liver, and skin.49 Mutations in the SKIV2L and TTC37 genes disrupt proteins that regulate intracellular mRNA turnover, causing multisystemic symptoms.50 Key characteristics of THES include severe, treatment-resistant diarrhea, malabsorption, and growth retardation, accompanied by nutritional deficiencies that manifest from infancy. Many patients also exhibit liver dysfunction, characterized by hepatomegaly, elevated liver enzyme levels, and, in some cases, cirrhosis-related splenomegaly. A hallmark feature of the syndrome is thin, brittle, and slow-growing hair, often associated with trichorexis nodosa, along with dry skin, eczematous rashes, and a texture prone to bruising. Recurrent infections due to immunodeficiency are common, while bone marrow suppression frequently results in anemia, thrombocytopenia, and leukopenia, further contributing to the clinical complexity of the disorder.49
Singleton-Merten Syndrome (SMS)
SMS is usually associated with GOF mutations in IFIH1. However, an atypical form caused by the DDX58 mutation has also been described.51 Characteristic features of SMS include dental abnormalities such as early tooth loss and enamel hypoplasia, which may provide important clues to the diagnosis. In addition, skeletal dysplasia, osteoporosis, and growth abnormalities in long bones have been observed in patients. Calcifications are common, especially in the hands and feet, and can lead to severe finger and spinal deformities.52 One of the hallmarks of SMS is vascular calcifications that progress with age. These calcifications, especially in the aorta and other heart valves, are common and often lead to serious cardiovascular complications.1 In addition, symptoms such as myalgia, skin rashes, growth retardation, and glaucoma have also been reported in cases.52
Interferon-Stimulated Gene 15 (ISIG15) Deficiency
ISG15 deficiency is an AR disorder caused by mutations in ISG15. This leads to impaired production of ISG15, a ubiquitin-like protein. ISG15 is an important molecule that enhances the immune response to viral infections through post-translational modification of intracellular proteins.53 Symptoms usually appear in the newborn or early childhood, including clinical signs such as fever, skin rashes, and recurrent lung infections. Patients become more susceptible to mycobacterial infections, especially tuberculosis.27 Seizures, psychomotor retardation, and developmental delays may also occur.54
Ubiquitin-Specific Protease 18 (USP18) Deficiency
USP18 deficiency results from heterozygous GOF mutations in the USP18 gene.53 The USP18 protein, in conjunction with ISG15, negatively regulates the type 1 IFN response.1 This condition typically manifests in the neonatal period, with symptoms such as severe skin rashes, fever, muscle weakness, and respiratory issues. Chronic CNS inflammation can lead to severe neurological symptoms, including seizures and developmental delays.1 It is termed “pseudo-TORCH syndrome” due to features like microcephaly, ventriculomegaly, and intracranial calcification without congenital infection.53
Retinal Vasculopathy with Cerebral Leukodystrophy (RVCL)
RVCL is an autosomal dominant (AD) disorder resulting from mutations in the TREX1 gene. This mutation results in the loss of function of the TREX1 protein, which is responsible for DNA fragmentation.55 Consequently, this leads to intracellular DNA accumulation and chronic inflammation, thereby triggering an autoimmune response that damages vascular and nerve tissues. Symptoms typically appear between the ages of 30 and 50 and include blurred vision, vision loss, cognitive impairment, headaches, seizures, and progressive dementia.56 Hypertension and renal failure have also been reported.55
X-linked Reticulate Pigmentary Disorder (XLRPD)
XLRPD occurs due to mutations in the POLA1 gene.57 Patients with XLRPD often experience photophobia and sometimes corneal opacities. Corneal thickening and reduced transparency may progress to the point where vision loss may occur. These symptoms usually appear early and may progress over time.58 One of the hallmarks of the disease is diffuse reticular hyperpigmentation that begins shortly after birth and affects the trunk, extremities, and face. Patients may also experience dry skin and eczematous rashes. Males with XLRPD often have additional manifestations such as recurrent respiratory and gastrointestinal infections, facial dysmorphia, corneal dyskeratosis, and hypohidrosis. Female carriers usually only exhibit cutaneous manifestations.58
Proteasome-associated Autoinflammatory Syndromes (PRAAS)/Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature (CANDLE)
PRAAS/CANDLE is an AR disease caused by mutations in the PSMB8, PSMB4, PSMB9, PSMB10, PSMA3, PSMB7, POMP, and PSMG2 genes.59 Patients with CANDLE syndrome frequently experience recurrent fevers from infancy, accompanied by neutrophilic dermatitis, characterized by purple plaques, swelling, painful rashes, and ulcerations on the skin.59 They exhibit significant facial and arm lipodystrophy, leading to both aesthetic and metabolic issues.60 Musculoskeletal symptoms such as arthralgia and muscle weakness are common.59 Due to chronic inflammation, visceral functions are negatively affected in these patients. Growth retardation, hepatosplenomegaly, lymphadenopathy, and hematologic disorders may be observed.60
DIAGNOSIS
Due to their rarity, type 1 interferonopathies necessitate thorough clinical evaluation, laboratory analysis, and genetic testing.1 A chronic and progressive inflammatory presentation may indicate interferonopathies, typically manifesting in childhood with unusual symptoms and resistance to conventional therapies.3 A wide range of symptoms are observed, including skin rashes, neurological findings due to central nervous system involvement, pulmonary symptoms, and musculoskeletal problems.5 Symptoms such as vasculitic skin changes, panniculitis, ILD, basal ganglia calcifications, neuromotor disorders, epilepsy, and recurrent fever are particularly common in these diseases.53 Since it has a Mendelian inheritance pattern, risk factors such as family history and consanguineous marriage are also taken into account in the diagnostic process.2
Diagnosing type 1 interferonopathies necessitates demonstrating an elevated type 1 interferon response; however, low blood levels present a challenge in measurement.1 The IFN signature method is an approach to assess ISG expression. Increased expression of genes such as IFIT1, IFI27, IFI44L, ISG15, RSAD2, and SIGLEC1 are important biomarkers supporting interferonopathies. Although a positive IFN signature is observed in most patients, results may be negative in some cases, especially AGS patients with RNASEH2B mutations.5 Positive results can also occur in viral infections or SLE, limiting specificity and risking false positives.2 Thus, combining the IFN signature with clinical findings and genetic analysis improves diagnostic accuracy.53
Genetic analysis is crucial for confirming the diagnosis and distinguishing interferonopathies from other diseases, as most gene mutations affect the interferon pathways. Identifying gene mutations like IFIH1, TREX1, TMEM173, and ADAR1 clarifies the diagnosis.3 Next-generation sequencing, such as whole-exome sequencing (WES) or whole-genome sequencing (WGS), is recommended for definitive diagnosis.61
In which patients should we suspect Type I interferonopathies?
Type I IFNs should be suspected in the presence of chronic autoinflammatory or autoimmune diseases resistant to conventional therapies, usually starting in early childhood, especially in the presence of unexplained neurologic or dermatologic symptoms.2 Findings such as livedo reticularis, ulceration or necrosis of the extremities, basal ganglia calcifications, treatment-resistant seizures, and unexplained musculoskeletal symptoms strongly support the possibility of Type I IFNs.5 In addition to these symptoms, factors such as family history, consanguinity, and genetic predisposition are also important clues that increase suspicion for the diagnosis.2
How should we approach the diagnosis in patients suspected of having interferonopathies?
The diagnosis of type 1 IFN starts with a high clinical suspicion, followed by a demonstration of an IFN response.1 For this, the IFN signature test is used, which measures ISG expression in the peripheral blood. Although the IFN signature is positive in most patients, it can be confused with other diseases owing to its low specificity.5 WES is preferred to clarify the diagnosis; however, it has limitations, such as its high cost and inability to detect intronic mutations. The most accurate diagnosis is made using WGS; however, it is rarely used because of its high cost.61 Therefore, an accurate diagnosis requires a multidisciplinary approach using clinical, laboratory, and genetic testing.
TREATMENT
Treating type 1 interferonopathies aims to alleviate symptoms and modulate the interferon response. Although no definitive cure exists, current strategies focus on suppressing IFN signaling. JAK inhibitors (ruxolitinib, tofacitinib, and baricitinib) block the JAK-STAT pathway, reducing inflammation and symptoms in conditions such as AGS. However, they are not fully curative and may not reverse lung involvement.62 Side effects include BK virus viremia and respiratory infections. Anti-inflammatory and immunosuppressive drugs (corticosteroids, methotrexate, mycophenolate mofetil) reduce general inflammation but do not directly target interferon signaling, thus offering a limited response.3
Anti-IFN antibodies (sifalimumab and anifrolumab) specifically neutralize interferon molecules and have shown promise in phase 2-3 trials.63 Reverse transcriptase inhibitors (abacavir, lamivudine, and zidovudine) provide short-term improvement in some patients but lack long-term efficacy.5 Gene therapy may offer future solutions but remains in the research phase.4 Due to their experimental nature, these treatments require monitoring by a multidisciplinary team and an individualized approach.
Acknowledgements
The authors would like to thank Kubra Ozturk, Feray Kaya, Lutfiye Koru, Zelal Aydin, Eda Nur Dizman, Hatice Kubra Dursun, and Merve Ozen Balci for their assistance in the follow-up of the patients with type with interferonopathy. Figure was created using BioRender.com. Parental permission was obtained for sharing the patient’s MRI photographs. Publication licenses for figure was obtained. Paperpal, an AI tool, was used for language editing.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429-40. https://doi.org/10.1038/nri3850
- Haşlak F, Kılıç Könte E, Aslan E, Şahin S, Kasapçopur Ö. Type I interferonopathies in childhood. Balkan Med J. 2023;40:165-74. https://doi.org/10.4274/balkanmedj.galenos.2023.2023-4-78
- Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. 2016;213:2527-38. https://doi.org/10.1084/jem.20161596
- Uggenti C, Lepelley A, Crow YJ. Self-awareness: nucleic acid-driven inflammation and the type I interferonopathies. Annu Rev Immunol. 2019;37:247-67. https://doi.org/10.1146/annurev-immunol-042718-041257
- Rice GI, Forte GMA, Szynkiewicz M, et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159-69. https://doi.org/10.1016/S1474-4422(13)70258-8
- Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell. 2018;34:361-78. https://doi.org/10.1016/j.ccell.2018.05.013
- López de Padilla CM, Niewold TB. The type I interferons: basic concepts and clinical relevance in immune-mediated inflammatory diseases. Gene. 2016;576:14-21. https://doi.org/10.1016/j.gene.2015.09.058
- Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873-86. https://doi.org/10.1016/j.cell.2007.10.017
- Briggs TA, Rice GI, Adib N, et al. Spondyloenchondrodysplasia due to mutations in ACP5: a comprehensive survey. J Clin Immunol. 2016;36:220-34. https://doi.org/10.1007/s10875-016-0252-y
- Desai R, Frazier AE, Durigon R, et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain. 2017;140:1595-610. https://doi.org/10.1093/brain/awx094
- Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380-4. https://doi.org/10.1038/nature12306
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674-8. https://doi.org/10.1038/nature07317
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786-91. https://doi.org/10.1126/science.1232458
- Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215:1287-99. https://doi.org/10.1084/jem.20180139
- Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol. 2014;5:342. https://doi.org/10.3389/fimmu.2014.00342
- Schlee M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology. 2013;218:1322-35. https://doi.org/10.1016/j.imbio.2013.06.007
- Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335-45. https://doi.org/10.1128/JVI.01080-07
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669-82. https://doi.org/10.1016/j.cell.2005.08.012
- Arimoto KI, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104:7500-5. https://doi.org/10.1073/pnas.0611551104
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727-40. https://doi.org/10.1016/j.molcel.2005.08.014
- Collins GA, Goldberg AL. The logic of the 26S proteasome. Cell. 2017;169:792-806. https://doi.org/10.1016/j.cell.2017.04.023
- d’Angelo DM, Di Filippo P, Breda L, Chiarelli F. Type I interferonopathies in children: an overview. Front Pediatr. 2021;9:631329. https://doi.org/10.3389/fped.2021.631329
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344-62. https://doi.org/10.1016/j.cell.2008.01.020
- Schlee M, Hartmann G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol. 2016;16:566-80. https://doi.org/10.1038/nri.2016.78
- Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491-6. https://doi.org/10.1038/ni921
- Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900-11. https://doi.org/10.1038/nri1226
- Bogunovic D, Boisson-Dupuis S, Casanova JL. ISG15: leading a double life as a secreted molecule. Exp Mol Med. 2013;45:e18. https://doi.org/10.1038/emm.2013.36
- Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol. 2006;16:79-87. https://doi.org/10.1016/j.tcb.2005.12.005
- Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75-86. https://doi.org/10.1111/j.1600-065X.2008.00737.x
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36-49. https://doi.org/10.1038/nri3581
- Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity. 2012;36:503-14. https://doi.org/10.1016/j.immuni.2012.03.013
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513-45. https://doi.org/10.1146/annurev-immunol-032713-120231
- McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87-103. https://doi.org/10.1038/nri3787
- Haslak F, Kilic H, Sahin S, et al. Children with type 1 interferonopathy: commonalities and diversities in a large patient cohort. The Journal of Rheumatology. 2024;52. https://doi.org/10.3899/jrheum.2024-0294
- Liu Y, Jesus AA, Marrero B, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507-18. https://doi.org/10.1056/NEJMoa1312625
- Jeremiah N, Neven B, Gentili M, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124:5516-20. https://doi.org/10.1172/JCI79100
- Frémond ML, Hadchouel A, Berteloot L, et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) among 21 patients. J Allergy Clin Immunol Pract. 2021;9:803-18.e11. https://doi.org/10.1016/j.jaip.2020.11.007
- Watkin LB, Jessen B, Wiszniewski W, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47:654-60. https://doi.org/10.1038/ng.3279
- Volpi S, Tsui J, Mariani M, et al. Type I interferon pathway activation in COPA syndrome. Clin Immunol. 2018;187:33-6. https://doi.org/10.1016/j.clim.2017.10.001
- Tsao BP, Grossman JM. Genetics and systemic lupus erythematosus. Curr Rheumatol Rep. 2001;3:183-90. https://doi.org/10.1007/s11926-001-0017-2
- Batu ED, Koşukcu C, Taşkıran E, et al. Whole exome sequencing in early-onset systemic lupus erythematosus. J Rheumatol. 2018;45:1671-9. https://doi.org/10.3899/jrheum.171358
- Alarcón-Riquelme ME. The genetics of systemic lupus erythematosus. J Autoimmun. 2005;25:46-8. https://doi.org/10.1016/j.jaut.2005.09.012
- Kisla Ekinci RM, Balci S, Bisgin A, Altintas DU, Yilmaz M. A homozygote TREX1 mutation in two siblings with different phenotypes: chilblains and cerebral vasculitis. Eur J Med Genet. 2017;60:690-4. https://doi.org/10.1016/j.ejmg.2017.09.004
- Bilginer Y, Düzova A, Topaloğlu R, et al. Three cases of spondyloenchondrodysplasia (SPENCD) with systemic lupus erythematosus: a case series and review of the literature. Lupus. 2016;25:760-5. https://doi.org/10.1177/0961203316629000
- Lausch E, Janecke A, Bros M, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. 2011;43:132-7. https://doi.org/10.1038/ng.749
- Melki I, Frémond ML. Type I interferonopathies: from a novel concept to targeted therapeutics. Curr Rheumatol Rep. 2020;22:32. https://doi.org/10.1007/s11926-020-00909-4
- Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911-20. https://doi.org/10.1056/NEJMoa1307361
- Caorsi R, Penco F, Grossi A, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. 2017;76:1648-56. https://doi.org/10.1136/annrheumdis-2016-210802
- Fabre A, Martinez-Vinson C, Goulet O, Badens C. Syndromic diarrhea/Tricho-hepato-enteric syndrome. Orphanet J Rare Dis. 2013;8:5. https://doi.org/10.1186/1750-1172-8-5
- Dweikat I, Sultan M, Maraqa N, Hindi T, Abu-Rmeileh S, Abu-Libdeh B. Tricho-hepato-enteric syndrome: a case of hemochromatosis with intractable diarrhea, dysmorphic features, and hair abnormality. Am J Med Genet A. 2007;143A:581-3. https://doi.org/10.1002/ajmg.a.31583
- Rutsch F, MacDougall M, Lu C, et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet. 2015;96:275-82. https://doi.org/10.1016/j.ajhg.2014.12.014
- Jang MA, Kim EK, Now H, et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am J Hum Genet. 2015;96:266-74. https://doi.org/10.1016/j.ajhg.2014.11.019
- Zhang X, Brann TW, Zhou M, et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol. 2011;186:4541-5. https://doi.org/10.4049/jimmunol.1003389
- Speer SD, Li Z, Buta S, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. 2016;7:11496. https://doi.org/10.1038/ncomms11496
- Richards A, van den Maagdenberg AMJM, Jen JC, et al. C-terminal truncations in human 3’-5’ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068-70. https://doi.org/10.1038/ng2082
- Wilms AE, de Boer I, Terwindt GM. Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic manifestations (RVCL-S): an update on basic science and clinical perspectives. Cereb Circ Cogn Behav. 2022;3:100046. https://doi.org/10.1016/j.cccb.2022.100046
- Starokadomskyy P, Gemelli T, Rios JJ, et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. 2016;17:495-504. https://doi.org/10.1038/ni.3409
- Chamoto K, Al-Habsi M, Honjo T. Role of PD-1 in immunity and diseases. Curr Top Microbiol Immunol. 2017;410:75-97. https://doi.org/10.1007/82_2017_67
- Kim H, Sanchez GAM, Goldbach-Mansky R. Insights from mendelian interferonopathies: comparison of CANDLE, SAVI with AGS, monogenic lupus. J Mol Med (Berl). 2016;94:1111-27. https://doi.org/10.1007/s00109-016-1465-5
- Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87:866-72. https://doi.org/10.1016/j.ajhg.2010.10.031
- Crow YJ. Type I interferonopathies: mendelian type I interferon up-regulation. Curr Opin Immunol. 2015;32:7-12. https://doi.org/10.1016/j.coi.2014.10.005
- Frémond ML, Rodero MP, Jeremiah N, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol. 2016;138:1752-5. https://doi.org/10.1016/j.jaci.2016.07.015
- Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol. 2022;22:471-83. https://doi.org/10.1038/s41577-021-00633-9
Copyright and license
Copyright © 2025 The author(s). This is an open-access article published by Aydın Pediatric Society under the terms of the Creative Commons Attribution License (CC BY) which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.