Abstract
Background: Osteopetrosis (OP) is a rare, severe inherited disorder of bone metabolism caused by impaired osteoclast function. The most severe form, malignant infantile osteopetrosis (MIOP), presents in early life with bone abnormalities, neurologic issues, and often hypocalcemia and carries a high mortality rate if left untreated. This study aimed to define the molecular spectrum and delineate genotype-phenotype correlations in a cohort of patients from a single pediatric hematology center.
Methods: We retrospectively reviewed the medical, laboratory, radiological, and genetic data of 12 children with OP followed between 2012 and 2022. Whole-exome sequencing was used for genetic analysis.
Results: The median age at diagnosis was three months. The most frequently mutated gene was TCIRG1 (n=7, 58.3%), followed by OSTM1 (n=3, 25%), and CLCN7 (n=2, 16.7%). A novel homozygous deletion in TCIRG1 was identified. A strong genotype-phenotype correlation was observed. Patients with TCIRG1 mutations predominantly presented with severe bone marrow failure and osteopetrorickets. In contrast, all patients with OSTM1 and CLCN7 mutations exhibited significant neurodegenerative changes. Two patients received hematopoietic stem cell transplantation (HSCT), with one survivor.
Conclusion: This study highlights the distinct clinical and genetic heterogeneity of MIOP. Our findings reinforce that the specific genetic mutation is critical for predicting the disease course—hematological complications for TCIRG1 versus primary neurodegeneration for OSTM1 and CLCN7. This genetic diagnosis is essential for counseling families and determining eligibility for curative therapies like HSCT.
Keywords: malignant infantile osteopetrosis, TCIRG1, OSTM1, CLCN7, genotype-phenotype correlation, marble bone disease
INTRODUCTION
Osteopetrosis (OP), also known as “marble bone disease,” is a group of rare, heritable skeletal disorders characterized by increased bone mass resulting from defective osteoclast development or function. This failure of bone resorption leads to generalized skeletal sclerosis, which paradoxically increases the risk of fractures.1-3 Consequently, overly dense bones may compress the cranial nerves, potentially resulting in irreversible nerve damage. Additionally, expansion of bone into the marrow cavities can lead to bone marrow failure.4
OP is classified into three clinical subtypes based on age of onset, severity, and inheritance patterns:
Autosomal recessive osteopetrosis (ARO) severe malignant infantile and intermediate form, and autosomal dominant osteopetrosis (ADO) late-onset form.2,5-7
Malignant infantile osteopetrosis (MIOP) represents the most severe form of osteopetrosis and is inherited in an autosomal recessive pattern. This form is genetically heterogeneous. Mutations in the TCIRG1 gene are the most common cause, accounting for over 50% of ARO cases.8-10 Biallelic mutations in T cell immunoregulator 1 (TCIRG1), chloride channel 7 (CLCN7), sequence nexin 10 (SNX10), osteopetrosis-associated transmembrane protein 1 (OSTM1), TNF receptor superfamily member 11a (TNFRSF11A/RANK), TNF Receptor Superfamily Member 11 (TNFSF11/RANKL), pleckstrin homology domain-containing family M -with RUN domain-member 1 (PLEKHM1), lead to ARO.11-15
The incidence of MIOP is 1/250,000 live births.8-10In individuals with MIOP, clinical manifestations typically appear shortly after birth and are associated with various systemic abnormalities.12,15 Common symptoms include recurrent infections, unusual bruising, and bleeding disorders.16Affected patients also experience frequent pathological fractures. Other symptoms include macrocephaly and hepatosplenomegaly, hearing loss, vision impairment, and hypocalcemia.17
Osteopetrosis is primarily diagnosed based on the presence of characteristic clinical and radiographic features. Genetic mutations can be identified in approximately 90% of patients with OP.2,11
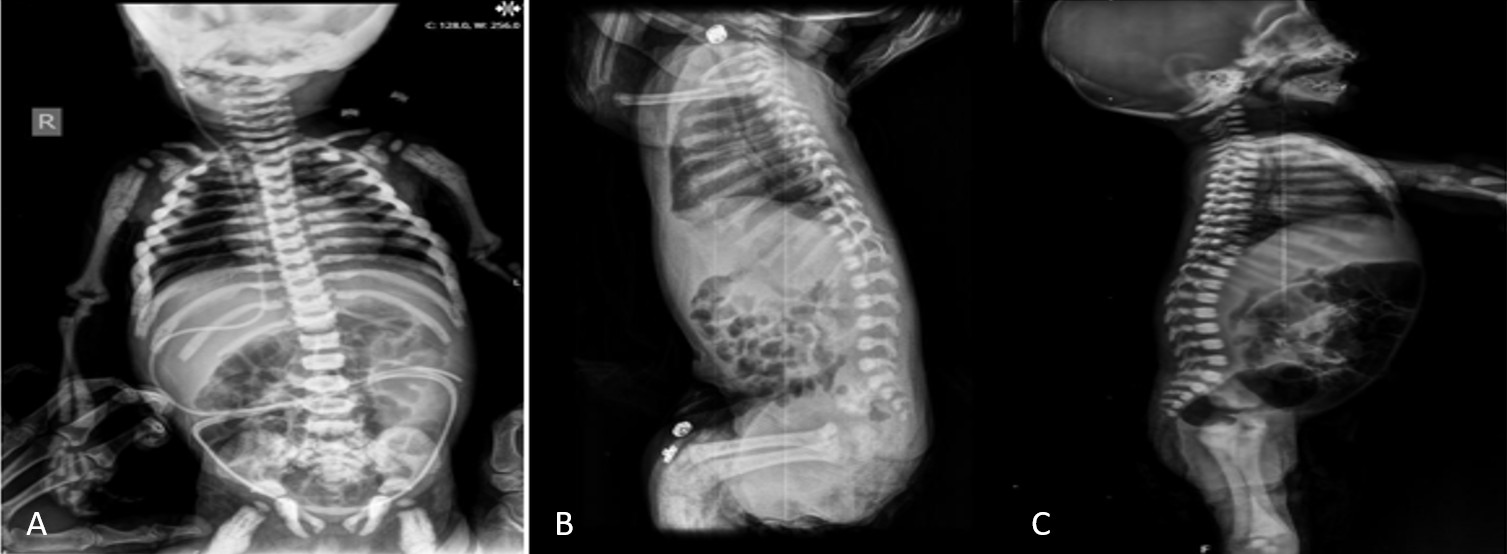
Radiographic examinations typically reveal generalized osteosclerosis throughout the skeleton, characterized by a “marble bone” appearance. The notable radiologic findings are: “Erlenmeyer flask” deformity, “bone-in-bone” or “endobone” appearance, and the “sandwich vertebrae” or “rugger jersey spine”.5
Genetic testing provides critical insights into prognosis and clinical correlates, which significantly influence management decisions.
HSCT is the treatment of choice for patients with OP. Clear indications for HSCT include progressive bone marrow failure or the imminent risk of vision loss due to optic nerve compression. However, HSCT is not suitable for all patients.18,19
The rarity of the disease, particularly in developing countries, often results in diagnostic delays that preclude timely intervention. This study aims to characterize the molecular spectrum and clinical features of 12 children with MIOP from a single center to better understand the genotype-phenotype correlations and highlight the diagnostic journey.
MATERIALS AND METHODS
Study design
This retrospective study included 12 children with osteopetrosis who were followed at a single pediatric hematology center between 2012 and 2022. Data on family history, physical examination findings, complete blood counts, serum biochemistry (25-hydroxyvitamin D, calcium, phosphorus, alkaline phosphatase, and parathyroid hormone), imaging results, and genetic analyses were retrieved from medical records. This study was approved by the Gaziantep University University Non-Interventional Clinical Research Ethics Committee (Date: 4 October 2023, Number: 2023/316), and written informed consent was obtained from the participants’ parents.
Genetic analysis
Genomic DNA was extracted from peripheral blood leukocytes of the patients and parents. Whole-exome sequencing libraries were prepared using the Twist Human Core Exome Kit and NovaSeq system (Illumina, USA). The variant interpretation followed a structured analysis algorithm. Sequence data were analyzed using SOPHIA DDM software. Variants were first filtered based on their frequency in the Genome Aggregation Database (gnomAD) to exclude common polymorphisms. The potential pathogenicity of remaining rare variants was assessed using multiple in silico prediction tools (e.g., LRT, CADD, EIGEN, BayesDel). Variants were then cross-referenced with public databases (ClinVar, HGMD) and literature sources before being classified according to the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines. Segregation analysis was performed using Sanger sequencing to confirm that variants were inherited from the parents.
Statistical analysis
Data were examined utilizing SPSS version 24 (IBM Corp., Armonk, NY). Quantitative (numerical) variables were displayed as mean, standard deviation, median, maximum, and minimum, whereas qualitative (categorical) variables were summarized in terms of frequency and percentage
RESULTS
Cohort characteristics
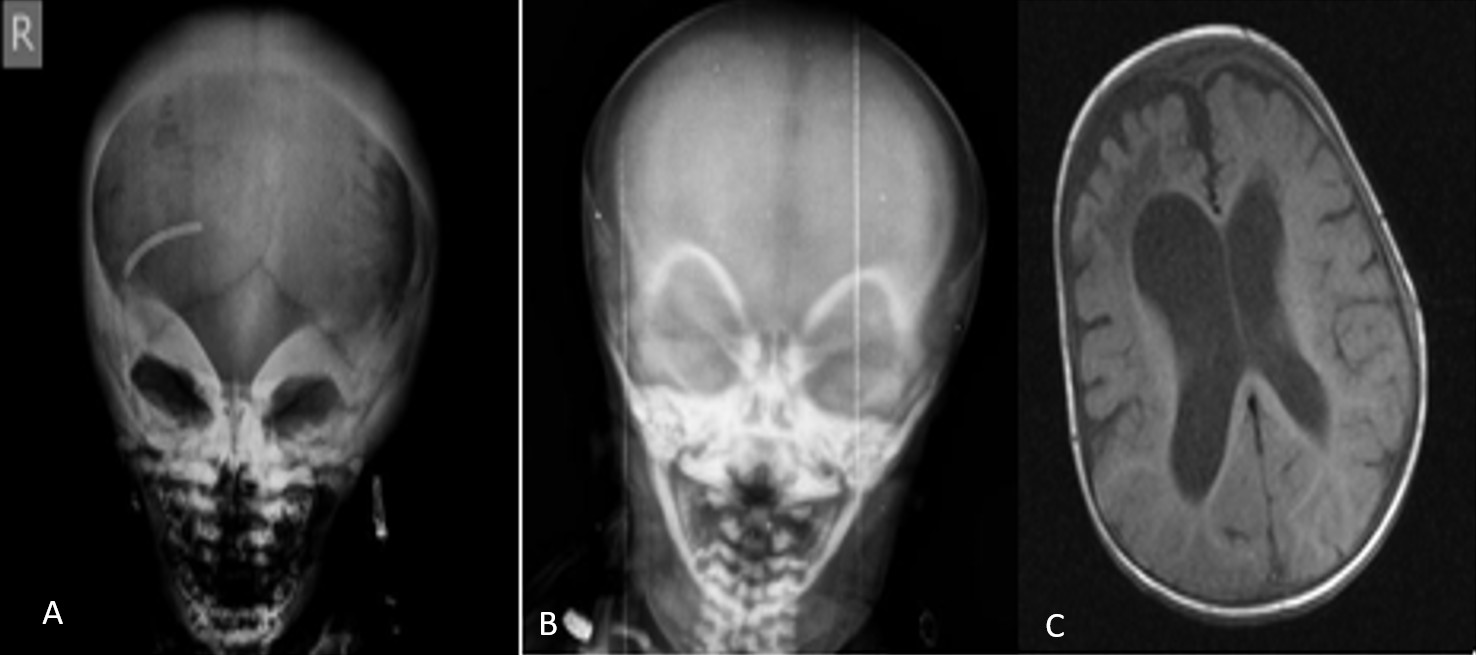
A total of 12 patients (7 female, 5 male) from ten distinct families were included. All patients were born to consanguineous parents. The median age at diagnosis was three months (range, 1 to 36.5 months). The most common presenting symptoms were nasal congestion and difficulty sucking (n=4) (Table 1). Radiographic assessments in all 12 patients revealed features typical of osteopetrosis, including diffuse bone sclerosis, thickened skull bones, a “sandwich vertebrae” appearance, and a “bone-within-bone” sign (Figure 1 and Figure 2).
| Table 1. Molecular findings of patients with osteopetrosis | |||||||
|---|---|---|---|---|---|---|---|
| Patient No. | Gene | Zygosity | Variant | Protein alteration | Location | Mutation type | ACMG classification |
| 1 | TCIRG1 | Homozygous | - | - | Exon 11-12 | Large deletion | Pathogenic |
| 2 | TCIRG1 | Homozygous | c.2236C>T (NM_006019.4) | p.Q746* p. Gln746* | Exon 18 | Nonsense | Pathogenic |
| 3 | TCIRG1 | Homozygous | c.2236+1G>A (NM_006019.4) | - | Intron 18 | Splice site | Pathogenic |
| 4 | TCIRG1 | Homozygous | c.936_963del (NM_006019.4) | p.Ser312ArgfsTer25 | Exon 9 | Indel | Likely pathogenic |
| 5 | TCIRG1 | Homozygous | c.2236+1G>A (NM_006019.4) | - | Intron 18 | Splice site | Pathogenic |
| 6 | TCIRG1 | Homozygous | - | - | Exon 11-13 | Large deletion | Pathogenic |
| 7 | TCIRG1 | Homozygous | - | - | Exon 11-13 | Large deletion | Pathogenic |
| 8 | OSTM1 | Homozygous | c.402+1G>T (NM_014028.3) | - | Intron 1 | Splice site | Likely pathogenic |
| 9 | OSTM1 | Homozygous | c.402+1G>T (NM_014028.3) | - | Intron 1 | Splice site | Likely pathogenic |
| 10 | OSTM1 | Homozygous | c.402+1G>T (NM_014028.3) | - | Intron 1 | Splice site | Likely pathogenic |
| 11 | CLCN7 | Homozygous | c.1576C>T (NM_001287) | p.(Arg526Trp) | Exon 17 | Missense | Pathogenic |
| 12 | CLCN7 | Homozygous | c.746C>G (NM_001287.5) | p.Pro249Arg | Exon 9 | Missense | Pathogenic |
Molecular findings
Genetic analysis identified pathogenic or likely pathogenic variants in all 12 patients (Table 2). The most frequently affected gene was TCIRG1 in seven patients (58.3%), followed by OSTM1 in three patients (25%), and CLCN7 in two patients (16.7%). All mutations were homozygous. A novel homozygous deletion in TCIRG1, c.936_963del (NM_006019.4), which causes a frameshift leading to a premature stop codon, was identified in Patient 4. This variant was absent from the gnomAD database and was classified as “likely pathogenic.” Other identified variants have been previously reported.
| Table 2. Clinical, laboratory and radiologic findings of patients with osteopetrosis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genetic mutation |
|
|
|
|||||||||
| F: Female, M:Male, HSCT: Hematopoietic stem cell transplantation, N/A: Not available | ||||||||||||
| Patient Number |
|
|
|
|
|
|
|
|
|
|
|
|
| Sex |
|
|
|
|
|
|
|
|
|
|
|
|
| Age at diagnosis (months) |
|
|
|
|
|
|
|
|
|
|
|
|
| Presenting symptom |
|
|
|
|
|
|
|
|
|
|
|
|
| Nystagmus |
|
|
|
|
|
|
|
|
|
|
|
|
| Strabismus |
|
|
|
|
|
|
|
|
|
|
|
|
| Vision loss |
|
|
|
|
|
|
|
|
|
|
|
|
| Pale optic disc |
|
|
|
|
|
|
|
|
|
|
|
|
| Hydrocephaly |
|
|
|
|
|
|
|
|
|
|
|
|
| Neurodegenerative changes |
|
|
|
|
|
|
|
|
|
|
|
|
| Hepatosplenomegaly |
|
|
|
|
|
|
|
|
|
|
|
|
| Bone fracture |
|
|
|
|
|
|
|
|
|
|
|
|
| Hearing loss |
|
|
|
|
|
|
|
|
|
|
|
|
| Seizures |
|
|
|
|
|
|
|
|
|
|
|
|
| Anemia |
|
|
|
|
|
|
|
|
|
|
|
|
| Thrombocytopenia |
|
|
|
|
|
|
|
|
|
|
|
|
| Leukocytosis |
|
|
|
|
|
|
|
|
|
|
|
|
| Hypocalcemia |
|
|
|
|
|
|
|
|
|
|
|
|
| HSCT |
|
|
|
|
|
|
|
|
|
|
|
|
| Death |
|
|
|
|
|
|
|
|
|
|
|
|
Clinical and laboratory findings by genotype
Clinical features varied significantly according to the underlying genetic defect (Table 1).
TCIRG1-related osteopetrosis (n=7)
This group comprised Patients 1, 2, 3, 4, 5, 6, and 7. Bone marrow failure was the predominant clinical feature. Six of the seven patients (85.7%) had anemia and/or thrombocytopenia at presentation or during follow-up. All patients in this group, except one, required transfusions, with six becoming transfusion-dependent. All seven patients had hepatosplenomegaly, indicative of extramedullary hematopoiesis. Vision loss was present in three patients, and pale optic discs were noted in six. Osteopetrorickets was diagnosed in six of the seven patients (85.7%). Neurodegenerative changes were not a feature in this group. At the time of the study, four patients in this group had died, two were alive under supportive care, and one was alive and well post-HSCT.
OSTM1-related osteopetrosis (n=3)
This group included Patients 8, 9, and 10, all of whom carried the same homozygous splice site mutation in OSTM1. These patients presented with a severe neurodegenerative phenotype. All three exhibited neurodegenerative changes on cranial imaging, including diffuse cerebral atrophy and a thin corpus callosum, and had a history of seizures. Two patients died during the study period.
CLCN7-related osteopetrosis (n=2)
Patients 11 and 12 had homozygous missense mutations in CLCN7. Both patients presented with a severe phenotype that included neurodegenerative changes. Patient 11 had hydrocephalus and seizures, while Patient 12 had vision loss and hepatosplenomegaly. Both patients in this group died.
Patient outcomes
Overall, eight of the twelve patients (66.7%) died during the study period. Four patients were still alive. Two patients underwent HSCT. Patient 3 (TCIRG1 mutation), who underwent HSCT, died from sepsis one month post-transplant. The other patient who received HSCT (Patient 5, TCIRG1 mutation) survived and remains in good health.
DISCUSSION
This study describes the clinical and genetic landscape of 12 children with MIOP from a single center, highlighting distinct genotype-phenotype correlations and identifying a novel pathogenic mutation. Our findings confirm that MIOP is a heterogeneous disorder where the underlying gene defect is a major determinant of the clinical course and prognosis.
In our cohort, TCIRG1 variants were identified as the likely causative gene for ARO in seven cases (58.3%), consistent with previous research indicating that TCIRG1 is the pathogenic gene in over 50% of ARO cases.8,10 In one patient (Patient 4), we identified a novel homozygous mutation in TCIRG1, while all other mutations had been previously reported.
The clinical presentation of our TCIRG1 patients was dominated by severe hematological complications and osteopetrorickets, which aligns with existing literature.20 This is biologically plausible, as the TCIRG1 gene encodes a crucial subunit of the proton pump necessary for osteoclasts to acidify the bone-resorption lacuna. Its dysfunction directly cripples bone resorption, leading to marrow cavity obliteration and subsequent bone marrow failure.
In stark contrast, all patients with OSTM1 and CLCN7 mutations exhibited a severe neurodegenerative phenotype. This finding highlights the broader physiological roles of the proteins encoded by these genes. Unlike TCIRG1, which is primarily expressed in osteoclasts, CLCN7 (a chloride channel) and OSTM1 (its beta-subunit) are also vital for the proper function of the endolysosomal system in various cell types, including neurons. Disruption of this pathway is thought to cause lysosomal storage defects within neurons, leading to the observed cerebral atrophy, seizures, and progressive neurological decline, which are absolute contraindications for HSCT.21,22
The prevalence of OSTM1 mutations in our cohort (25%) is significantly higher than the ~5% reported in broader studies, which may suggest a founder effect within our population, especially since all three affected patients carried the same homozygous mutation.12,15
Mutations in the CLCN7 gene are the second most common cause of ARO, accounting for 17% of cases.2,5,20,22,23 In the current study, we identified CLCN7 mutations in 16.7% of the patients, which aligns with previous reports. However, this mutation was the third most prevalent in our cohort. Our patients with biallelic CLCN7 mutations also exhibited neurodegenerative features, consistent with reports that approximately half of such cases involve neurological decline.21
For many patients with MIOP, hematopoietic stem cell transplantation (HSCT) is the only curative treatment option, particularly in cases of progressive bone marrow failure or optic nerve compression. However, the suitability of HSCT is highly dependent on the specific underlying genetic mutation and the patient’s clinical presentation, making early and accurate genetic diagnosis crucial for management decisions. Absolute contraindications include extrinsic osteoclast defects caused by RANKL mutations and the severe neurodegenerative forms of OP associated with all known OSTM1 mutations and about half of biallelic CLCN7 mutations. In osteopetrosis, the conditioning regimen must strike a delicate balance between achieving sufficient myeloablation and immunosuppression while minimizing regimen-related toxicity.18,19
TCIRG1 mutations, who underwent HSCT, with starkly different outcomes. Patient 5 received an HSCT and is alive and in good health. Patient 3, however, died from sepsis one month post-transplant.
While a detailed analysis is limited by the small sample size, this highlights the critical challenges of HSCT in this population. Patients with MIOP are often malnourished and have recurrent infections, making them highly susceptible to regimen-related toxicity and post-transplant complications like severe infections. The choice of conditioning regimen, donor source, and the timing of the transplant before irreversible organ damage occurs are paramount for success. These cases underscore the need for early genetic diagnosis to identify suitable candidates (i.e., those without primary neurodegeneration) and to proceed with HSCT under optimal clinical conditions to minimize the risk of fatal complications.
CONCLUSION
This study underscores the significant clinical and genetic heterogeneity of Malignant Infantile Osteopetrosis (MIOP) and reinforces the critical role of genotype-phenotype correlations in clinical management. Our findings confirm that hematological and skeletal symptoms should raise suspicion for TCIRG1-related osteopetrosis, while early and severe neurological decline is characteristic of forms caused by OSTM1 and some CLCN7 mutations. The identification of a novel pathogenic TCIRG1 mutation contributes to the growing catalog of variants for this disease. Ultimately, a timely and comprehensive assessment that combines clinical, radiological, and genetic data is essential for accurate diagnosis, prognostication, and guiding crucial therapeutic decisions, particularly regarding the feasibility and timing of curative therapies like HSCT.
Ethical approval
This study has been approved by the Gaziantep University University Non-Interventional Clinical Research Ethics Committee (approval date 04.10.2023, number 2023/307). Written informed consent was obtained from the participants.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. 2009;4:5. https://doi.org/10.1186/1750-1172-4-5
- Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9:522-36. https://doi.org/10.1038/nrendo.2013.137
- Bailey JR, Tapscott DC. Osteopetrosis. [Updated 2023 Apr 24]. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025. Available at: https://www.ncbi.nlm.nih.gov/books/NBK557529/
- Dozier TS, Duncan IM, Klein AJ, Lambert PR, Key LL. Otologic manifestations of malignant osteopetrosis. Otol Neurotol. 2005;26:762-6. https://doi.org/10.1097/01.mao.0000178139.27472.8d
- Wu CC, Econs MJ, DiMeglio LA, et al. Diagnosis and management of osteopetrosis: consensus guidelines from the osteopetrosis working group. J Clin Endocrinol Metab. 2017;102:3111-3123. https://doi.org/10.1210/jc.2017-01127
- Balemans W, Van Wesenbeeck L, Van Hul W. A clinical and molecular overview of the human osteopetroses. Calcif Tissue Int. 2005;77:263-74. https://doi.org/10.1007/s00223-005-0027-6
- Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42:19-29. https://doi.org/10.1016/j.bone.2007.08.029
- Bliznetz EA, Tverskaya SM, Zinchenko RA, et al. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: the unique splice site mutation in TCIRG1 gene spread by the founder effect. Eur J Hum Genet. 2009;17:664-72. https://doi.org/10.1038/ejhg.2008.234
- Anderson SL, Jalas C, Fedick A, et al. A founder mutation in the TCIRG1 gene causes osteopetrosis in the Ashkenazi Jewish population. Clin Genet. 2015;88:74-9. https://doi.org/10.1111/cge.12448
- Zlotogora J. Autosomal recessive diseases among Palestinian Arabs. J Med Genet. 1997;34:765-6. https://doi.org/10.1136/jmg.34.9.765
- Palagano E, Menale C, Sobacchi C, Villa A. Genetics of osteopetrosis. Curr Osteoporos Rep. 2018;16:13-25. https://doi.org/10.1007/s11914-018-0415-2
- Pillai NR, Aggarwal A, Orchard P. Phenotype-autosomal recessive osteopetrosis. Bone. 2022;165:116577. https://doi.org/10.1016/j.bone.2022.116577
- Frattini A, Orchard PJ, Sobacchi C, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. 2000;25:343-6. https://doi.org/10.1038/77131
- Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23:447-51. https://doi.org/10.1038/70563
- Villa A, Guerrini MM, Cassani B, Pangrazio A, Sobacchi C. Infantile malignant, autosomal recessive osteopetrosis: the rich and the poor. Calcif Tissue Int. 2009;84:1-12. https://doi.org/10.1007/s00223-008-9196-4
- Coudert AE, de Vernejoul MC, Muraca M, Del Fattore A. Osteopetrosis and its relevance for the discovery of new functions associated with the skeleton. Int J Endocrinol. 2015;2015:372156. https://doi.org/10.1155/2015/372156
- Kornak U, Kasper D, Bösl MR, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205-15. https://doi.org/10.1016/s0092-8674(01)00206-9
- Schulz A, Moshous D. Hematopoietic stem cell transplantation, a curative approach in infantile osteopetrosis. Bone. 2023;167:116634. https://doi.org/10.1016/j.bone.2022.116634
- Schulz A, Moushous D, Steward CG, Villa A, Sobacchi C. Osteopetrosis: consensus guidelines for diagnosis, therapy and follow-up. 2015. Available at: https://esid.org/wp-content/uploads/2024/03/00_OP_Guidelines_V3.pdf
- Kaplan FS, August CS, Fallon MD, Gannon F, Haddad JG. Osteopetrorickets: the paradox of plenty. Pathophysiology and treatment. Clin Orthop Relat Res. 1993;(294):64-78. https://doi.org/10.1097/00003086-199309000-00008
- Pangrazio A, Caldana ME, Lo Iacono N, et al. Autosomal recessive osteopetrosis: report of 41 novel mutations in the TCIRG1 gene and diagnostic implications. Osteoporos Int. 2012;23:2713-8. https://doi.org/10.1007/s00198-011-1878-5
- Sobacchi C, Frattini A, Orchard P, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet. 2001;10:1767-73. https://doi.org/10.1093/hmg/10.17.1767
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for molecular pathology. Genet Med. 2015;17:405-24. https://doi.org/10.1038/gim.2015.30
Copyright and license
Copyright © 2025 The author(s). This is an open-access article published by Aydın Pediatric Society under the terms of the Creative Commons Attribution License (CC BY) which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.