Abstract
Objective: Ebstein’s anomaly is a rare congenital heart defect featuring a varied spectrum of clinical presentation. Anatomical variability in the malformation, together with a frequent association with other cardiac defects and arrhythmias, can complicate diagnosis and long-term management.
Methods: Data from a total of 26 patients with Ebstein’s anomaly followed up at two university clinics between 2009 and 2025 were analyzed retrospectively. Cases diagnosed from the prenatal period through adolescent period were included.
Results: The cohort consisted of 26 patients; 69.2% (18/26) were males with a median diagnostic age of 8 days. Diagnoses were established prenatally 15.4% (4/26), neonatally 50.0% (13/26), or during later childhood and adolescence 34.6% (9/26). Diagnosis was made during the index NICU admission in 34.6% (9/26) of patients and based on referral for murmur 26.9% (7/26) or cyanosis 7.7% (2/26). Common associated anomalies included atrial-level shunts 61.5% (16/26), mitral regurgitation 23.1% (6/26), and ventricular septal defects 15.4% (4/26). Arrhythmias were documented in 23.1% (6/21) of the cohort; 42.3% (11/26) underwent catheter-based procedures, and 30.8% (8/26) required surgery (median age at first surgery, 8.4 months), with 15.4%(4/26) undergoing reoperation. Over a median 5-year follow-up, mortality was 15.4% (4/26), with no perioperative deaths. Among prenatally diagnosed cases (n=4), three deaths occurred (3/4, 75%).
Conclusions: The clinical spectrum of Ebstein’s anomaly is exceptionally broad. Our findings show frequent left heart involvement and a substantial need for catheter-based and surgical interventions, supporting the importance of individualized assessment and follow-up across the disease course.
Keywords: Ebstein’s anomaly, prenatal diagnosis, congenital heart defect
INTRODUCTION
A rarely seen congenital cardiac malformation, Ebstein’s anomaly is often attributed to abnormal delamination of the tricuspid valve leaflets during embryogenesis, and it is defined by apical displacement of the septal and posterior leaflets.1 Incidence is relatively low; according to the most recent counts, the estimated rate stood at 0.2–0.7 per 10,000 live births, so the condition accounts for 0.3–0.6% of all congenital heart defects.2,3
Aberrant displacement and adherence of the leaflets is thought to split the right ventricle into functionally different atrialized and ventricular components.4 In doing so, the malformation affects the tricuspid valve, the right ventricle, and the electrical conduction system alike.5 Clinical presentation therefore varies widely. At one end, intrauterine demise and profound neonatal cyanosis; at the other, incidental diagnosis in adults who remain asymptomatic for decades. For the most part, such variation traces back to the severity of the anatomical derangement itself; the degree of leaflet displacement besides right ventricular impairment largely sets the clinical course.1
In neonates, cyanosis and congestive heart failure, as well as pronounced cardiomegaly are the usual findings. Older children tend to show right ventricular failure instead, whereas adolescents and adults more often come to clinical attention because of arrhythmias.6 Of note, the anomaly frequently coexists with an atrial septal defect (ASD) or patent foramen ovale (PFO); they have been reported in more than 80% of patients and may predispose them to paradoxical embolism.7
Since the condition can be highly variable in its clinical presentation and accompanying anomalies can be frequent, Ebstein’s anomaly should be managed on an individualized patient-by-patient basis. We therefore aimed to describe the clinical, electrocardiographic, and echocardiographic findings, the burden of catheter-based and surgical interventions, and the outcomes of 26 patients followed up from fetal life through adolescent period at two university clinics.
MATERIAL and METHOD
Our study followed a retrospective cohort design. Patients diagnosed with Ebstein’s anomaly were included. All had been managed at the Pediatric Cardiology Clinics of the Faculties of Medicine of Necmettin Erbakan University and Selçuk University between 2009 and 2025. Patients were identified by searching the institutional electronic health record database for relevant diagnostic codes and reviewing clinic records. The study protocol received approval from the Necmettin Erbakan University Faculty of Medicine Ethics Committee (Ethics approval number: 2025/5792), which waived the need for individual patient consent due to the study’s retrospective nature. All data were handled in a de-identified/anonymized manner in accordance with institutional policies. De-identified data underlying the findings of this study are available from the corresponding author upon reasonable request.
The primary inclusion criterion was a definitive diagnosis of Ebstein’s anomaly confirmed by transthoracic echocardiography with availability of sufficient clinical and imaging data for phenotypic characterization and outcome assessment. Patients with secondary tricuspid regurgitation without morphological features of Ebstein’s anomaly or with records insufficient to confirm the diagnosis were excluded. Patients with incomplete clinical or follow-up data precluding outcome assessment were excluded from the analysis.
Diagnostic confirmation was based on the Mayo Clinic criteria, which specify a septal tricuspid valve leaflet displacement toward the apex of ≥0.8 cm/m2 body surface area from the mitral valve anterior leaflet insertion point during systole.8 All echocardiograms were reviewed retrospectively, and anatomical severity was categorized according to the Carpentier classification (types A-D) based on echocardiographic morphology, including the functional right ventricular volume, the extent of atrialized right ventricle, and the mobility/tethering of the anterior tricuspid leaflet. All echocardiograms were independently reviewed by two pediatric cardiologists; disagreements were resolved by consensus. Associated cardiac lesions (e.g., ASD/PFO, Ventricular Septal Defect (VSD), left-sided lesions) were recorded from echocardiography and relevant operative/interventional reports. Valvular insufficiency severity was classified as mild, moderate, or severe according to established Doppler and color flow imaging guidelines. Tricuspid regurgitation severity was graded using an integrative Doppler approach (color Doppler jet characteristics and supportive qualitative/quantitative parameters when available) in accordance with standard echocardiography recommendations.9
We evaluated demographic and clinical data, including age at diagnosis, gender, date of last follow-up, presenting signs and symptoms (e.g., murmur, cyanosis, heart failure), and clinical outcomes, including mortality; electrocardiographic findings, echocardiographic data, details of any cardiac catheterization, electrophysiological study with ablation, or surgical intervention, including the type of procedure, age at intervention, and any complications and long-term follow-up outcomes. Given the retrospective design, some variables were incompletely documented in a small subset of patients; missing data were reported explicitly in table footnotes. Analyses were performed using a complete-case approach without imputation. Unless otherwise specified, proportions were calculated using n=26 as the denominator; subgroup proportions (e.g., surgically treated patients) used the corresponding subgroup denominator.
Statistical analysis
Statistical analysis was performed with IBM SPSS Statistics for Windows v.22.0 (IBM Corp., Armonk, NY, USA). Descriptive statistics are presented as median (minimum–maximum), frequency distributions, and percentages. Continuous variables were summarized as median (min–max) given the small sample size and non-normal distributions. Given the small sample size, analyses were primarily descriptive and inferential subgroup comparisons were not performed.
RESULTS
The study cohort comprised 26 patients with Ebstein’s anomaly. The timing of diagnosis varied: 15.4% (4/26) of cases were identified prenatally through fetal echocardiography, 50% (13/26) during the neonatal period, 19.2% (5/26) during childhood, and 15.4% (4/26) during adolescence. The median age at diagnosis was 8 days (range: 1 day–18 years), with a male predominance 69.2% (18/26). Among surviving patients, the median follow-up duration was 5 years (range: 1–16 years). As for the pathway to diagnosis, 34.6% (9/26) were diagnosed during the index NICU hospitalization, whereas 26.9% (7/26) were referred for evaluation of a murmur and 7.7% (2/26) for cyanosis. Other reasons included recurrent pneumonia, referral based on an outside medical report, and missing/unknown presentation. All patients had tricuspid regurgitation, which was classified as mild in 23.1% (6/26) of patients, moderate in 57.7% (15/26), and severe in 19.2% (5/26).
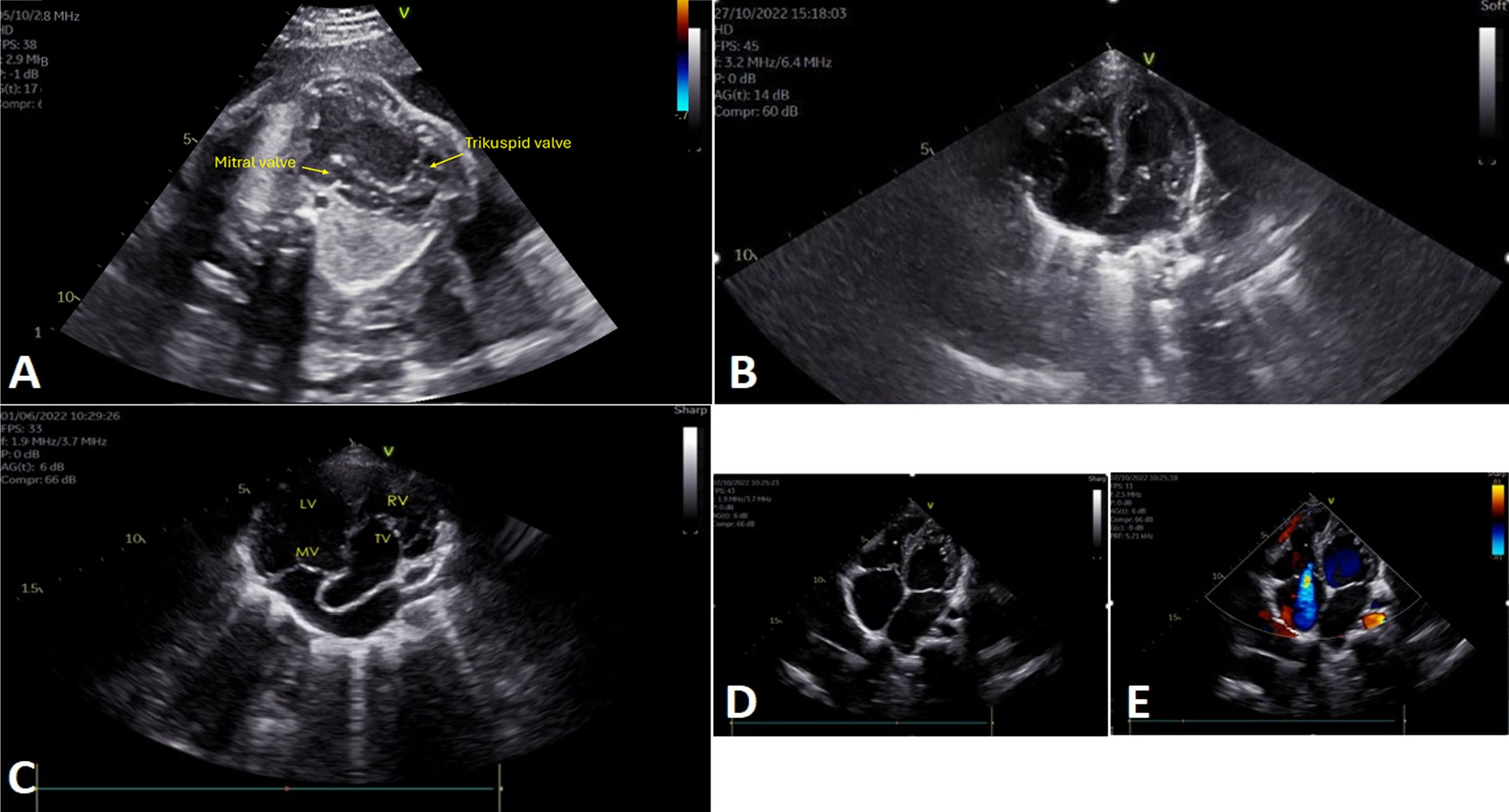
Anatomical subtyping according to the Carpentier classification for Ebstein’s anomaly distributed the cohort as follows: Type A in 38.5% (10/26), Type B in 26.9% (7/26), Type C in 19.2% (5/26), and Type D in 15.4% (4/26). Detailed findings are presented in Table 1, with representative echocardiographic images shown in Figure 1. The most common associated anomalies were an ASD/PFO 61.5% (16/26), mild-to-moderate mitral regurgitation 23.1% (6/26), and a ventricular septal defect (VSD) 15.4% (4/26). Further findings included pulmonary hypoplasia/atresia 11.5% (3/26) and corrected transposition of the great arteries 11.5% (3/26). Left heart involvement, encompassing VSD, bicuspid aortic valve, and mitral regurgitation, was present in 50% (13/26) of patients. One patient also had left ventricular noncompaction cardiomyopathy.
| *Includes recurrent pneumonia (n=1), outside medical report (n=1), and missing/unknown presentation (n=2) | |
| Table 1. Clinical features of the patients | |
| Features |
|
| Gender | |
| Female |
|
| Male |
|
| Age at diagnosis (median) (min-max) |
|
| Diagnosis period | |
| Fetal |
|
| Neonatal |
|
| Child |
|
| Adolescent |
|
| Presenting status / reason for referral | |
| Murmur |
|
| Cyanosis |
|
| Admission to the neonatal intensive care unit |
|
| Others/Unknown* |
|
| Prenatal diagnosis on fetal echocardiography |
|
| Tricuspid regurgitation degree | |
| Mild |
|
| Moderate |
|
| Severe |
|
| Carpentier classification | |
| Type A |
|
| Type B |
|
| Type C |
|
| Type D |
|
| Follow-up duration |
|
Arrhythmic disorders were documented in 23.1% (6/26) of patients. Three patients with Wolff–Parkinson–White (WPW) syndrome underwent successful catheter ablation. Among the remaining cases, one patient had supraventricular ectopy and one had ventricular ectopy. One patient with congenitally corrected transposition of the great arteries (ccTGA) developed complete atrioventricular block postoperatively and required permanent pacemaker implantation. Cardiac catheterization and interventional procedures were performed in 42.3% (11/26) of patients. The main interventions included diagnostic angiography 11.5% (3/26) and electrophysiological study with ablation 11.5% (3/26), with other procedures performed as clinically indicated. Another procedure was transcatheter ASD device closure (n=1).
Cardiac surgery was required in 30.8% (8/26) of patients, with a median age at the first intervention of 8.4 months (range: 4 days–25 years). Secondary surgical procedures were necessary in 15.4% (4/26) of cases. As for surgical timing, 11.5% (3/26) underwent surgical intervention during the neonatal period, and 23.1% (6/26) underwent surgery within the first year of life. The principal surgical techniques included tricuspid annuloplasty, cone reconstruction, Blalock–Taussig shunt placement, and double-switch procedures. Other surgical procedures were VSD closure (n=2), patent ductus arteriosus (PDA) ligation (n=2), and pulmonary artery banding (n=1) (Table 2). During a median follow-up of 5 years (range, 1–16 years), four patients died (15.4%, 4/26). Two of the four had been diagnosed prenatally with massive cardiomegaly and died in utero. A third patient died in the neonatal intensive care unit from sepsis and multiple organ failure. The last patient was a neonate who had been born to a mother with a known history of substance use after unmonitored pregnancy. The infant had severe tricuspid regurgitation and pulmonary hypoplasia and died within 24 hours of birth.
| *Some patients underwent more than one procedure. | |
| Table 2. Associated anomalies, interventional and surgical procedures performed | |
| Cardiac anomaly (n=26) |
|
| ASD/PFO |
|
| Congenitally Corrected Transposition of the great arteries |
|
| VSD |
|
| Pulmonary Hypoplasia/Atresia |
|
| Aortic regurgitation |
|
| Mild-to-moderate mitral regurgitation |
|
| Right aortic arch |
|
| Coarctation of the aorta |
|
| Patent Ductus Arteriosus |
|
| Bicuspid Aortic Valve |
|
| Left Ventricular Noncompaction |
|
| Catheter-based and electrophysiologic procedures (n=26) | |
| Diagnostic Angiography |
|
| Ablation |
|
| Pulmonary Balloon Valvuloplasty |
|
| Aortic Balloon Angioplasty |
|
| Transcatheter ASD closure |
|
| Right pulmonary artery (RPA) stenting |
|
| Balloon Atrial Septostomy |
|
| Surgery protocols | |
| Patients with Surgery |
|
| Age at first surgery(month) |
|
| Patients with second surgery |
|
| Surgery Type (procedures)* | |
| Tricuspid Valvuloplasty |
|
| Pulmonary Artery Banding |
|
| Double Switch |
|
| Cone Reconstruction |
|
| Coarctation repair (patch aortoplasty) |
|
| PDA ligation |
|
In adolescent patients, on the other hand, diagnoses were made at ages 13, 14, 16, and 18 years. Three of these patients had mild tricuspid regurgitation and were clinically stable during the follow-up period. At 16 years of age, the fourth patient had been diagnosed with corrected transposition of the great arteries; tricuspid valve annuloplasty and pulmonary artery banding were carried out at 25 years. No perioperative deaths were recorded.
DISCUSSION
In our experience at two university clinics, Ebstein’s anomaly can be said to show considerable variability from fetal life through adolescent period. Research providing longitudinal data covering the full developmental range is still rather limited. Our series of 26 patients, including cases detected prenatally, has therefore documented the frequency of left-heart involvement, burden of catheter-based and surgical interventions, and outcomes at each developmental stage. Most of the findings appear to be in concordance with earlier cohorts, but our fetal-to-adolescent coverage within one dataset can add to what has been reported so far in the current literature. In their study, Adıgüzel et al. reported that the mean age at diagnosis was 1.5 years (range: 1 day–24 years), with 51.9% males and 7.6% prenatal diagnoses.10 In the multicenter study by Kapusta et al., the median age at diagnosis was under 30 days, median follow-up was 86 months, and 81% of patients survived.11 Perinatal diagnoses were more frequent in our series, which could be attributed to the fact that our tertiary-level centers made use of fetal echocardiographic screening on a regular basis.
According to current evidence, patients who are diagnosed at early stage often tend to present with cyanosis and heart failure, but those diagnosed later generally have tachyarrhythmias or remain entirely asymptomatic.12 Oxenius et al. found that murmur was the most frequent finding at 33%, which was followed by cyanosis at 29%, with arrhythmia in 5%.13 Likewise, in the Adıgüzel series, cyanosis stood at 29.1% and murmur at 34.2%; 44.3% of their patients were asymptomatic.10 Our findings were similar; murmur was the most common reason for referral, and most diagnoses were made during the neonatal period.
Ebstein’s anomaly is known to be accompanied by a range of other structural defects in the human heart. For example, ASD or PFO have been reported in 80–94% of such cases.14 Oxenius et al. found that ASD/PFO were present in 79% of their patients, along with pulmonary outflow tract obstructions observed in 17% and less frequent anomalies like left ventricular noncompaction and VSD.13 Comparable figures were noted in the Adıgüzel series, where ASD/PFO were detected in 57% of patients, mitral regurgitation in 25.3%, pulmonary stenosis or atresia in 17.7%, as well as VSD in 16.5%.10 Tricuspid regurgitation in that cohort was found in more than three-quarters of cases (75.9%) and it varied in severity: mild in 11.7%, moderate in 38.3%, and severe in 50%. Our own findings appear to corroborate with literature reports.
In light of recent observations in relevant works, Ebstein’s anomaly may well extend beyond isolated right heart pathologies due to the fact that myocardial or valvular abnormalities of the left heart have been observed in as many as 39% of patients. These left heart lesions include left ventricular myocardial changes 17.9% resembling noncompaction, left ventricular dysfunction 43%, VSD 8%, bicuspid aortic valve 8%, mitral valve prolapse 15%, and mitral valve dysplasia 4%.5 Adıgüzel et al. documented left heart lesions in 58% of their cases, including VSD 16.5%, mitral regurgitation 25.3%, mitral prolapse 8.9%, aortic coarctation 3.8%, bicuspid aortic valve 2.5%, and one case of left ventricular noncompaction.10 Our patient cohort showed similarly high rates of left heart involvement, including VSD 15.4% (4/26), mitral regurgitation 23.1% (6/26), coarctation of the aorta 3.8%(1/26), bicuspid aortic valve 11.5% (3/26), and noncompaction 3.8% (1/26). The findings point to the need for thorough left ventricular assessment during echocardiographic evaluation.
Around one-third of patients with Ebstein’s anomaly exhibit atrioventricular conduction system abnormalities, with WPW syndrome reported in 5–25% of cases.14 Delhaas et al. documented a 17% arrhythmia prevalence in children with this condition, predominantly supraventricular tachycardia, with 15% showing pre-excitation patterns.15 Karagöz et al. identified arrhythmias in 30.3% of their patients, with ablation performed in 31%; the majority of these arrhythmias 82% were associated with accessory pathways.6 Our study detected WPW syndrome in 11.5% of patients, which seems to be in corroboration with the ranges published in previous research. Another consideration is that Ebstein’s anomaly also tends to require a high rate of invasive procedures; in our series, for example 42.3% of patients were found to show the need to undergo interventional catheterization. Relatively higher but comparable intervention rates have been reported by Geerdink et al. (60%) and Adıgüzel et al. (75%)4, which were diagnostic angiography (52.5%), electrophysiological ablation (35.6%), and palliative surgery (11.9%). All of them can be considered to showcase the heavy disease burden over the course of a patient’s lifetime.
Surgical intervention becomes necessary in cases with right heart enlargement, progressive ventricular dysfunction, or substantial valve insufficiency.16 Contemporary surgical techniques allow for palliation of patients with early neonatal findings through tricuspid valvuloplasty, with optional atrial septal fenestration and surgical shunts. Subsequently, selected cases with pulmonary atresia or stenosis may undergo staged procedures, including one-and-a-half ventricle repair or Fontan-type operations.17 The study by Oxenius et al.13 found that 79% of patients required invasive treatment, with 50% undergoing at least one surgical intervention at a median age of 9.1 years (range 0.1–16.5 years). Adıgüzel et al. reported cardiac surgery in 31.6% of patients, with an average age at first surgery of 6.5 years (range: 4 days–29 years).10 In their study, second surgical interventions were performed in 28% (n = 7) of patients, whereas this rate was 15% in our study.
The long-term prognosis for fetuses diagnosed with Ebstein’s anomaly remains poor, with fetal mortality reaching 20%; the overall perinatal mortality rate approximates 45%.18 In the study by Adıgüzel et al.10, with an average follow-up of 5.3 years, mortality occurred in 10.1% of patients at a median age of 25 days. Causes of death included heart failure, sudden cardiac death, necrotizing enterocolitis, and renal failure. Our study’s perinatal mortality was 75% (3/4) among prenatally diagnosed cases, which may reflect the small cohort size and the presence of massive cardiomegaly in these patients.
Our study included a single case with documented maternal heroin use during pregnancy. This single observation is worth noting, as the literature reports associations between Ebstein’s anomaly and certain maternal exposures (e.g., lithium and benzodiazepines), although evidence remains limited and inconsistent.19,20 However, it does not establish a teratogenic link and should be interpreted with caution. The infant in our cohort presented with a severe form of the anomaly and died on the first postnatal day, which we report as a hypothesis-generating observation rather than evidence of causality; potential confounding (including polysubstance exposure and incomplete prenatal care) cannot be excluded. Further systematically designed studies are needed to evaluate whether any association exists between maternal substance exposures and Ebstein’s anomaly.
CONCLUSION
The clinical picture of Ebstein’s anomaly was varied in our cohort where diagnoses spanned from fetal life to adolescent period. A high frequency of neonatal diagnosis, often in an intensive care setting for the most severe presentations, suggests earlier recognition of severe cases compared with historical reports. This rare defect is rather complex and can affect more than the tricuspid valve, with left heart involvement as defined in this study observed in half our patients and a frequent need for catheter-based or surgical interventions during follow-up. Over the long term, treatment outcomes may depend on how well disease management is individualized on a patient-by-patient basis, and the plan could involve early anatomical diagnosis, close monitoring for arrhythmias, and a plan for the timing and type of intervention.
Limitations
Our cohort size was naturally small since Ebstein’s anomaly is a rare condition, which left little room for meaningful subgroup comparisons. Analyses were thus kept descriptive; comparisons by diagnostic period, Carpentier type, or left-heart involvement were not attempted, because they would have been underpowered and potentially misleading. Some variables like diagnostic pathway and maternal exposure history were incompletely documented in the medical record, and a complete-case approach without imputation was employed. Management strategies also evolved over the long study period, and our findings reflect the experience of two tertiary centers, which can limit how far the results can be generalized.
Ethical approval
This study was approved by the Necmettin Erbakan University Faculty of Medicine Ethics Committee (Decision/Protocol No: 2025/5792). Ethics committee approval and informed consent were not required for this study.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflict of interest
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors declare that this study received no funding.
Generative AI statement
The authors declare that no generative AI or AI-assisted technologies were used in the writing or preparation of this study.
References
- Demir N, Akçoral A, Koyuncuoğlu M, Bülbül Y. Prenatal ultrasonographic diagnosis of Ebstein anomaly. Perinatoloji Dergisi. 1994;2:221-4.
- Sharma N, Lalnunnem TJ, Nandwani M, Santa SA, Synrang BW. Ebstein anomaly with pregnancy: a rare case. J Reprod Infertil. 2018;19:119-22.
- Fuchs MM, Connolly HM. Ebstein anomaly in the adult patient. Cardiol Clin. 2020;38:353-63. https://doi.org/10.1016/j.ccl.2020.04.004
- Geerdink LM, Kapusta L. Dealing with Ebstein’s anomaly. Cardiol Young. 2014;24:191-200. https://doi.org/10.1017/S1047951113001169
- Attenhofer Jost CH, Tan NY, Hassan A, et al. Sudden death in patients with Ebstein anomaly. Eur Heart J. 2018;39:1970-7a. https://doi.org/10.1093/eurheartj/ehx794
- Karagöz T, Ertuğrul İ, Aypar E, et al. Two decades of experience on ablation in children with Ebstein’s anomaly. Cardiol Young. 2022;32:437-43. https://doi.org/10.1017/S1047951121002353
- Brown ML, Dearani JA, Danielson GK, et al. The outcomes of operations for 539 patients with Ebstein anomaly. J Thorac Cardiovasc Surg. 2008;135:1120-36, 1136.e1-7. https://doi.org/10.1016/j.jtcvs.2008.02.034
- O’Leary PW. Ebstein’s malformation and tricuspid valve diseases. In: Eidem BW, O’Leary PW, Cetta F, editors. Echocardiography in Pediatric and Adult Congenital Heart Disease. 2nd ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2015: 146-165.
- Lancellotti P, Tribouilloy C, Hagendorff A, et al. Recommendations for the echocardiographic assessment of native valvular regurgitation: an executive summary from the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2013;14:611-44. https://doi.org/10.1093/ehjci/jet105
- Adıgüzel A, Aypar E, Karagöz T, et al. Ebstein’s anomaly in children and young adults: clinical features, arrhythmia, surgical management, and factors affecting arrhythmia and mortality. Cardiol Young. 2025;35:38-45. https://doi.org/10.1017/S1047951124025599
- Kapusta L, Eveleigh RM, Poulino SE, et al. Ebstein’s anomaly: factors associated with death in childhood and adolescence: a multi-centre, long-term study. Eur Heart J. 2007;28:2661-6. https://doi.org/10.1093/eurheartj/ehm398
- Chang YM, Wang JK, Chiu SN, et al. Clinical spectrum and long-term outcome of Ebstein’s anomaly based on a 26-year experience in an Asian cohort. Eur J Pediatr. 2009;168:685-90. https://doi.org/10.1007/s00431-008-0820-0
- Oxenius A, Attenhofer Jost CH, Prêtre R, et al. Management and outcome of Ebstein’s anomaly in children. Cardiol Young. 2013;23:27-34. https://doi.org/10.1017/S1047951112000224
- Khositseth A, Danielson GK, Dearani JA, Munger TM, Porter CJ. Supraventricular tachyarrhythmias in Ebstein anomaly: management and outcome. J Thorac Cardiovasc Surg. 2004;128:826-33. https://doi.org/10.1016/j.jtcvs.2004.02.012
- Delhaas T, Sarvaas GJDM, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol. 2010;31:229-33. https://doi.org/10.1007/s00246-009-9590-3
- Yuan SM. Ebstein’s Anomaly: genetics, clinical manifestations, and management. Pediatr Neonatol. 2017;58:211-215. https://doi.org/10.1016/j.pedneo.2016.08.004
- Knott-Craig CJ, Goldberg SP, Overholt ED, Colvin EV, Kirklin JK. Repair of neonates and young infants with Ebstein’s anomaly and related disorders. Ann Thorac Surg. 2007;84:587-92. https://doi.org/10.1016/j.athoracsur.2007.03.061
- Freud LR, Escobar-Diaz MC, Kalish BT, et al. Outcomes and predictors of perinatal mortality in fetuses with Ebstein anomaly or tricuspid valve dysplasia in the current era: a multicenter study. Circulation. 2015;132:481-9. https://doi.org/10.1161/CIRCULATIONAHA.115.015839
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA. 1994;271:146-50.
- Correa-Villaseñor A, Ferencz C, Neill CA, Wilson PD, Boughman JA. Ebstein’s malformation of the tricuspid valve: genetic and environmental factors. Teratology. 1994;50:137-47. https://doi.org/10.1002/tera.1420500208
Copyright and license
Copyright © 2026 The author(s). This is an open-access article published by Aydın Pediatric Society under the terms of the Creative Commons Attribution License (CC BY) which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.