Introduction
Dear Editor,
Pediatric antiphospholipid syndrome (APS) remains a challenging diagnosis due to its rarity, heterogeneous presentation, and frequent overlap with other autoimmune or inflammatory disorders. Unlike adults, children often present with non-thrombotic manifestations, and features such as chronic thrombocytopenia or valvular involvement may easily be misinterpreted as more common pediatric conditions.1,2 Early recognition is critical to preventing long-term complications, yet delays frequently occur because APS is not initially suspected.
With this letter, we aim to highlight a diagnostically challenging case in which a child initially labeled as having immune thrombocytopenic purpura (ITP) and later acute rheumatic fever (ARF) was ultimately diagnosed with APS. This case underscores the importance of maintaining a broad differential diagnosis in children with persistent cytopenias and unexplained cardiac findings, and it demonstrates how early recognition of APS-related features can change the entire trajectory of care.
CASE REPORT
A 13-year-old male first presented at the age of seven with petechial purpura on his trunk. Laboratory investigations revealed thrombocytopenia, leading to an initial diagnosis of ITP. He was treated with intravenous immunoglobulin (IVIG) and steroids; however, no sustained response was achieved. Eltrombopag was also ineffective, and repeated bone marrow evaluations ruled out myelodysplasia or malignancy.
At the age of nine, the patient developed fever, chest pain, and arthralgia. The chest pain was non-positional and unaffected by movement. Laboratory findings showed elevated troponin (40 ng/L) and brain natriuretic peptide (BNP) (>400 pg/mL), raising suspicion of multisystem inflammatory syndrome in children (MIS-C)-related myocarditis, and treatment with IVIG and steroids was initiated.
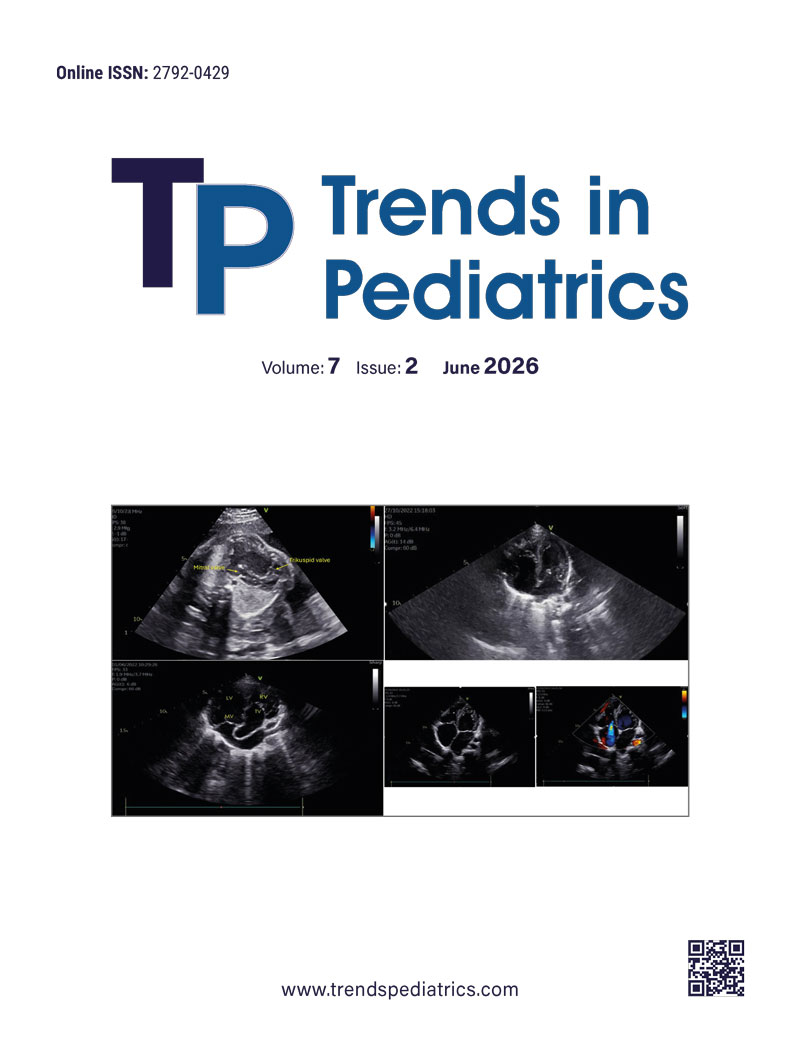
During follow-up, echocardiography revealed an eccentric jet flow, mitral valve thickening, and mitral regurgitation, but no thrombus was detected. Given the patient’s previous history of arthralgia, the condition was initially considered acute rheumatic fever (ARF), and secondary penicillin prophylaxis was initiated. However, upon referral to our hospital, the patient’s history of thrombocytopenia raised suspicion of APS, prompting further evaluation.
Laboratory findings revealed positive antiphospholipid antibodies, including anti-cardiolipin IgG (81.3 GPL/mL), beta-2 glycoprotein IgG (52.6 RU/mL), and a highly positive lupus anticoagulant test (LA ratio: 2.24, dRVVT: 86 sec), while anti-cardiolipin IgM and beta-2 glycoprotein IgM were negative. His autoantibody profile, including ANA, anti-dsDNA, and anti-Sm were negative.
Based on the presence of antiphospholipid antibody positivity, thrombocytopenia, and mitral valve thickening, the patient was classified as having APS according to the 2023 ACR/EULAR APS classification criteria. In this context, the prior history of arthralgia—without additional supportive features or laboratory evidence suggestive of acute rheumatic fever—was not considered sufficient to support a diagnosis of ARF, and APS was therefore favored. Treatment with intravenous immunoglobulin and corticosteroids was subsequently initiated in a stepwise manner. However, as the steroid dose was tapered, he developed severe thrombocytopenia (<10,000/mm³) and mucosal bleeding, necessitating rituximab therapy (2 doses of 500 mg/m² at 2-week intervals).
By the first month of follow-up, the platelet count improved to 223,000/mm³. By the third month, persistent antiphospholipid antibody positivity led to the initiation of low-dose acetylsalicylic acid (ASA). In the sixth month, the platelet count increased to 308,000/mm³ without any new symptoms. As a result, rituximab was not repeated.
At the one-year follow-up, the patient remained clinically stable, with no recurrence of symptoms. He continued low-dose ASA during follow-up.
In the present case, APS was considered primary, as there was no clinical, serological, or immunological evidence of an underlying systemic autoimmune connective tissue disease. In particular, features suggestive of juvenile systemic lupus erythematosus, such as hypocomplementemia, anti-dsDNA or anti-Sm antibody positivity, renal or neuropsychiatric involvement, or persistent inflammatory markers, were absent during follow-up.3
Mitral valve abnormalities are a well-recognized but often underestimated feature of APS. On echocardiography, APS-related valvular involvement is typically characterized by non-inflammatory leaflet thickening and small, sterile vegetations, whereas ARF is associated with inflammatory carditis, showing leaflet edema, commissural fusion, and chordal involvement. Notably, the “Sapporo” classification criteria for APS primarily focus on thrombotic events, which might not fully capture the spectrum of APS in pediatric patients.4 However, the 2023 ACR/EULAR classification criteria provide a more comprehensive framework for identifying APS, particularly in cases with non-thrombotic presentations.5 In our case, the valvular involvement was initially misdiagnosed as ARF. However, APS-related valvular disease often presents with thrombotic vegetations and valvular dysfunction, resembling other valvular disorders such as ARF. This diagnostic challenge was overcome through antiphospholipid antibody testing, leading to the correct diagnosis.
Although chronic ITP is the most common cause of persistent thrombocytopenia, APS should be considered in patients who are unresponsive to standard immunomodulatory therapies.6 In cases where IVIG, steroids, and thrombopoietin receptor agonists (e.g., eltrombopag) fail, recurrent episodes of thrombocytopenia may indicate an underlying systemic autoimmune disorder such as APS. Therefore, routine screening for antiphospholipid antibodies should be considered in patients with chronic, treatment-resistant thrombocytopenia. Moreover, transient aPL positivity can also be observed in healthy individuals, necessitating cautious interpretation of results. For instance, low-to-moderate lupus anticoagulant positivity or isolated positivity of a single antiphospholipid antibody is not uncommon and may not always indicate APS.7 The ACR/EULAR-recommended scoring system places greater emphasis on persistent lupus anticoagulant positivity in repeated tests, reflecting its stronger association with APS-related complications. 5 Furthermore, strongly positive lupus anticoagulant results or simultaneous positivity for three different antiphospholipid antibodies are linked to a higher risk of APS-related complications, reinforcing the need for a holistic risk assessment in clinical practice.7
The management of APS is a dynamic process that requires continuous reassessment of thrombotic and bleeding risks. In cases of active bleeding or severe thrombocytopenia, temporary discontinuation of antithrombotic therapy may be warranted to mitigate hemorrhagic risk.6 However, as platelet levels recover or in cases of mild thrombocytopenia, the balance shifts toward an increased thromboembolic risk, necessitating careful consideration of prophylactic anticoagulation. Individualized risk assessment, considering both bleeding and thrombotic tendencies, is essential to optimize management in APS patients with fluctuating platelet counts.
Rituximab is a monoclonal antibody targeting CD20, a surface protein expressed on B lymphocytes.8 By depleting B cells, it reduces autoantibody production and modulates immune responses, making it an effective treatment for various autoimmune and hematologic conditions.9 In APS, where B-cell dysregulation plays a role in persistent autoantibody production and immune-mediated thrombocytopenia, rituximab offers a targeted therapeutic approach, especially in refractory cases.10
Rituximab has been utilized in cases of life-threatening organ involvement, refractory thrombocytopenia, and, according to some case series, for the treatment of APS-related cutaneous ulcers and cognitive dysfunction.11-13
This case highlights the diverse clinical manifestations of pediatric APS, which can extend beyond thrombotic events to include hematologic abnormalities, such as thrombocytopenia, or cardiac involvement, such as mitral valve vegetations. Given the rarity and complexity of pediatric APS, early recognition and a comprehensive, multidisciplinary approach are crucial to minimizing morbidity and improving outcomes.
The 2023 ACR/EULAR classification criteria offer a more structured framework for APS diagnosis, yet the variability in pediatric presentations highlights the need for greater clinical awareness and a refined diagnostic approach. Moving forward, international collaborative efforts are crucial to establishing pediatric-specific classification criteria and standardized treatment protocols, ultimately improving disease management and long-term outcomes.
Conflict of interest
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors declare that this study received no funding.
Generative AI statement
The authors declare that no generative AI or AI-assisted technologies were used in the writing or preparation of this study.
References
- Grace RF, Lambert MP. An update on pediatric ITP: differentiating primary ITP, IPD, and PID. Blood. 2022;140:542-55. https://doi.org/10.1182/blood.2020006480
- Islabão AG, Trindade VC, da Mota LMH, Andrade DCO, Silva CA. Managing antiphospholipid syndrome in children and adolescents: current and future prospects. Paediatr Drugs. 2022;24:13-27. https://doi.org/10.1007/s40272-021-00484-w
- Sahin S, Adrovic A, Barut K, et al. Juvenile systemic lupus erythematosus in Turkey: demographic, clinical and laboratory features with disease activity and outcome. Lupus. 2018;27:514-9. https://doi.org/10.1177/0961203317747717
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306. https://doi.org/10.1111/j.1538-7836.2006.01753.x
- Barbhaiya M, Zuily S, Naden R, et al. The 2023 ACR/eular antiphospholipid syndrome classification criteria. Arthritis Rheumatol. 2023;75:1687-702. https://doi.org/10.1002/art.42624
- Tomasello R, Giordano G, Romano F, et al. Immune thrombocytopenia in antiphospholipid syndrome: is it primary or secondary? Biomedicines. 2021;9:1170. https://doi.org/10.3390/biomedicines9091170
- Aguiar CL, Soybilgic A, Avcin T, Myones BL. Pediatric antiphospholipid syndrome. Curr Rheumatol Rep. 2015;17:27. https://doi.org/10.1007/s11926-015-0504-5
- Leandro M, Isenberg DA. Rituximab - The first twenty years. Lupus. 2021;30:371-7. https://doi.org/10.1177/0961203320982668
- Cohen H, Cuadrado MJ, Erkan D, et al. 16th International Congress on Antiphospholipid Antibodies Task Force report on antiphospholipid syndrome treatment trends. Lupus. 2020;29:1571-93. https://doi.org/10.1177/0961203320950461
- Petri M. Antiphospholipid syndrome. Transl Res. 2020;225:70-81. https://doi.org/10.1016/j.trsl.2020.04.006
- Erkan D. Expert perspective: management of microvascular and catastrophic antiphospholipid syndrome. Arthritis Rheumatol. 2021;73:1780-90. https://doi.org/10.1002/art.41891
- Erkan D, Vega J, Ramón G, Kozora E, Lockshin MD. A pilot open-label phase II trial of rituximab for non-criteria manifestations of antiphospholipid syndrome. Arthritis Rheum. 2013;65:464-71. https://doi.org/10.1002/art.37759
- Nageswara Rao AA, Arteaga GM, Reed AM, Gloor JM, Rodriguez V. Rituximab for successful management of probable pediatric catastrophic antiphospholipid syndrome. Pediatr Blood Cancer. 2009;52:536-8. https://doi.org/10.1002/pbc.21878
Copyright and license
Copyright © 2026 The author(s). This is an open-access article published by Aydın Pediatric Society under the terms of the Creative Commons Attribution License (CC BY) which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.