
To the Editor,
Autosomal recessive mutations in the RAB27A gene (located on chromosome 15q21 and encoding Rab27a) cause Griscelli syndrome type 2 (GS-2).1 GS-2 is characterized by hypopigmentation and early-onset, potentially fatal hemophagocytic lymphohistiocytosis (HLH). Rab27a interaction with Munc 13-4 is fundamental for the secretion of lytic granules. Cytotoxic T lymphocytes and natural killer cells lacking Rab27a exhibit defective cytotoxicity and target cell killing.1 Besides, the Rab27a-melanophilin-myosin VA complex is required for the release of melanosomes from melanocytes. Therefore, GS-2 patients also show partial oculocutaneous albinism with silver-grey hair and large pigment aggregates of melanosomes within melanocytes in hair shafts. Recently, GS-2 cases, both late onset and sine albinism, have been reported in the literature.2-7 Here, we present a pediatric patient diagnosed with late-onset GS-2 without hypopigmentation.
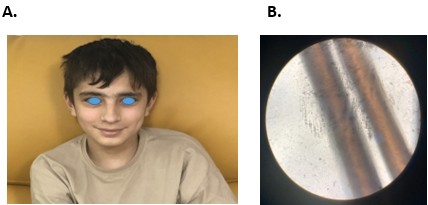
A fourteen-year-old boy was admitted to the pediatric emergency department in his hometown due to a fever. Physical examination did not reveal pathologic findings except splenomegaly and mild oropharyngeal hyperemia. After receiving ceftriaxone, he had a sudden loss of consciousness and cardiac arrest. After resuscitation, he was intubated. Cranial imaging was performed and didn’t reveal any pathologic findings supporting intracranial infections. Laboratory tests revealed haemolytic anemia haemoglobin 1.3 g/dL; direct Coombs test, 3+) and thrombocytopenia (platelet count, 21,000/mm³). Because of his thrombocytopenia and clinical deterioration, lumbar punction was not performed. He received erythrocyte and platelet transfusions, inotropes, intravenous immunoglobulin (IVIG), and pulse corticosteroid therapy. On follow-up, he was referred to our pediatric intensive care unit (PICU) in need of plasmapheresis and continuous renal replacement therapy. On PICU admission, physical examination revealed splenomegaly, nonspecific rashes, and extensive mucosal bleeding. Aside from severe acute kidney injury findings requiring continuous venovenous hemodiafiltration, laboratory tests revealed anemia, thrombocytopenia, low haptoglobin, and elevated ferritin, triglyceride levels, positive direct Coombs, elevated liver enzymes, and bone marrow hemophagocytosis supporting the diagnosis of HLH (Table 1).8 Because of his complicated clinical course, an immunology consultation was planned. Background history revealed that he was born to non-consanguineous parents, and over the last two years, he has been followed by splenomegaly and intermittent pancytopenia/bicytopenia. Neither he nor his siblings had fever episodes or neurological symptoms. Myelodysplastic syndrome, paroxysmal nocturnal hemoglobinuria, Gaucher disease, and Niemann-Pick disease were excluded. We evaluated our patient for possible causes for HLH (e.g., infections, malignancy, immunodeficiency) via immunologic work-up, viral serology, bacterial cultures, radiologic imaging, and bone marrow aspiration. Laboratory assessment revealed EBV viremia, low IgM levels, and low absolute lymphocyte subset counts (Table 1). He received rituximab, anakinra, IVIG, and steroid treatments. Exome sequencing (ES) was performed and identified compound heterozygous [c.488G>T (p.Ser163Ile), and c.-3A>C] variants of unknown significance in the RAB27A gene (NM_183235.3). Sanger sequencing validated that the c.488G>T (p. Ser163Ile) variant was maternally and the c.-3A>C variant was paternally segregated and found to be in trans configuration. Contrary to expectations, he did not have hypopigmentation (Figure 1). After discharge from PICU, he was treated according to HLH 94:2018 consensus recommendations.9 Eight months after HLH treatment and remission, he was transplanted from his 10/10 HLA-matched elder sister (who had a heterozygous c.-3A>C variant of unknown significance in the RAB27A gene). His post-transplant course was uncomplicated. Neutrophil engraftment was achieved on day +14, and no profound thrombocytopenia was seen. Chimerism analysis of the peripheral blood showed full donor chimerism on day +21. He is now well at post-transplant 10 months with 100% chimerism.
| ALT: Alanine Aminotransferase; AST: Aspartate Aminotransferase; EBV: Epstein-Barr virus; NK: Natural Killer cells; PCR: Polymerase Chain Reaction. Bold values indicate abnormal laboratory levels. |
||
| Table 1. Laboratory and immunologic assessments of the patient who previously received IVIG and steroid | ||
|
|
|
|
| Haemoglobin (g/dL) |
|
|
| Platelets (/mm3) |
|
|
| Absolute neutrophile count (/mm3) |
|
|
| Absolute lymphocyte count (/mm3) |
|
|
| Ferritin (ng/mL) |
|
|
| Triglyceride level (mg/dL) |
|
|
| Direct Coombs |
|
|
| AST (U/L) |
|
|
| ALT (U/L) |
|
|
| Haptoglobin (g/L) |
|
|
| Fibrinogen (mg/dl) |
|
|
| IgG (mg/dl) |
|
|
| IgA (mg/dl) |
|
|
| IgM (mg/dl) |
|
|
| T. Ig E (IU/mL) |
|
|
| Isohemagglutinin titer |
|
|
| EBV PCR (Viral load) (copy/ml) |
|
|
| CD3+T cells (%, /mm3) |
|
|
| CD3+CD4+ T cells (%, /mm3) |
|
|
| CD3+CD8+ T cells (%, /mm3) |
|
|
| CD19+ B cells (%, /mm3) |
|
|
| CD3-CD16+CD56+ NK cells (%, /mm3) |
|
|
Recently, a comprehensive dataset of 149 GS-2 patients diagnosed up to now was published.7 According to this study, 31 patients (21%) did not have any signs of albinism, and 16 patients in the cohort (11%) developed initial symptoms at age 10 years or over (late-onset). The most common presenting feature in patients with late-onset was systemic HLH (n = 10, 63%). Sixty-nine percent of these patients (11/16) did not exhibit features of partial albinism. In addition, 5 of 8 patients with viral infections at HLH presentation had EBV viremia. Our patient’s clinical features were also consistent with the findings in this publication.
In conclusion, our case also highlights that hypopigmentation can be absent in GS-2 and should not preclude the diagnosis. RAB27A mutations should be investigated in patients with suspected HLH disease without hypopigmentation.
Ethical approval
Both parents and patient provided the written informed consent.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Stephan Ehla RAM, Genevie’ ve de Saint Basile. Genetic diseases predisposing to HLH. In: Kathleen E. Sullivan ERS, editors. Stiehm’s immune deficiencies inborn errors of immunity. 2nd ed. United Kingdom: Academic Press; 2020. p. 549-72. https://doi.org/10.1016/B978-0-12-816768-7.00023-5
- Cetica V, Hackmann Y, Grieve S, et al. Patients with Griscelli syndrome and normal pigmentation identify RAB27A mutations that selectively disrupt MUNC13-4 binding. J Allergy Clin Immunol. 2015;135:1310-8.e1. https://doi.org/10.1016/j.jaci.2014.08.039
- Tesi B, Rascon J, Chiang SCC, et al. A RAB27A 5’ untranslated region structural variant associated with late-onset hemophagocytic lymphohistiocytosis and normal pigmentation. J Allergy Clin Immunol. 2018;142:317-21.e8. https://doi.org/10.1016/j.jaci.2018.02.031
- Ohishi Y, Ammann S, Ziaee V, et al. Griscelli syndrome type 2 sine albinism: unraveling differential RAB27A effector engagement. Front Immunol. 2020;11:612977. https://doi.org/10.3389/fimmu.2020.612977
- Woodward KE, Shah RM, Benseler S, et al. Considering immunologic and genetic evaluation for HLH in neuroinflammation: a case of Griscelli syndrome type 2 with neurological symptoms and a lack of albinism. Pediatr Blood Cancer. 2020;67:e28312. https://doi.org/10.1002/pbc.28312
- Zondag TCE, Torralba-Raga L, Van Laar JAM, et al. Novel RAB27A variant associated with late-onset hemophagocytic lymphohistiocytosis alters effector protein binding. J Clin Immunol. 2022;42:1685-95. https://doi.org/10.1007/s10875-022-01315-4
- Maimaris J, Roa-Bautista A, Sohail M, et al. Griscelli syndrome type 2: comprehensive analysis of 149 new and previously described patients with RAB27A deficiency. J Clin Immunol. 2024;45:50. https://doi.org/10.1007/s10875-024-01842-2
- Coffey K, Minnicozzi S. Hemophagocytic lymphohistiocytosis: an update in diagnostics, criteria, and treatment considerations. Curr Opin Pediatr. 2025;37:619-24. https://doi.org/10.1097/MOP.0000000000001508
- Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. 2018;6:1508-17. https://doi.org/10.1016/j.jaip.2018.05.031
Copyright and license
Copyright © 2026 The author(s). This is an open-access article published by Aydın Pediatric Society under the terms of the Creative Commons Attribution License (CC BY) which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.
